O(N) Ab-initio framework for materials under extreme conditons
In order to overcome the critical cubic-scaling bottleneck with respect to system size, much research in the past two decades has been devoted to the development of linear-scaling solution strategies for DFT. Rather than calculate the orthonormal Kohn-Sham orbitals, these techniques directly determine the quantities of interest with linear-scaling cost by exploiting the nearsightedness of matter. Though these efforts have yielded significant advances, there are a number of limitations. In particular, the accuracy and stability of linear-scaling methods remain ongoing concerns due to the need for additional computational parameters, subtleties in determining sufficient numbers and/or centers of localized orbitals, limitations of underlying basis sets, and calculation of accurate atomic forces, as required for structural relaxation and molecular dynamics simulations. In addition, efficient large-scale parallelization poses a significant challenge due to complex communications patterns and load balancing issues. Finally, and perhaps most importantly, the assumption of a band gap in the electronic structure makes these methods inapplicable to metallic systems.
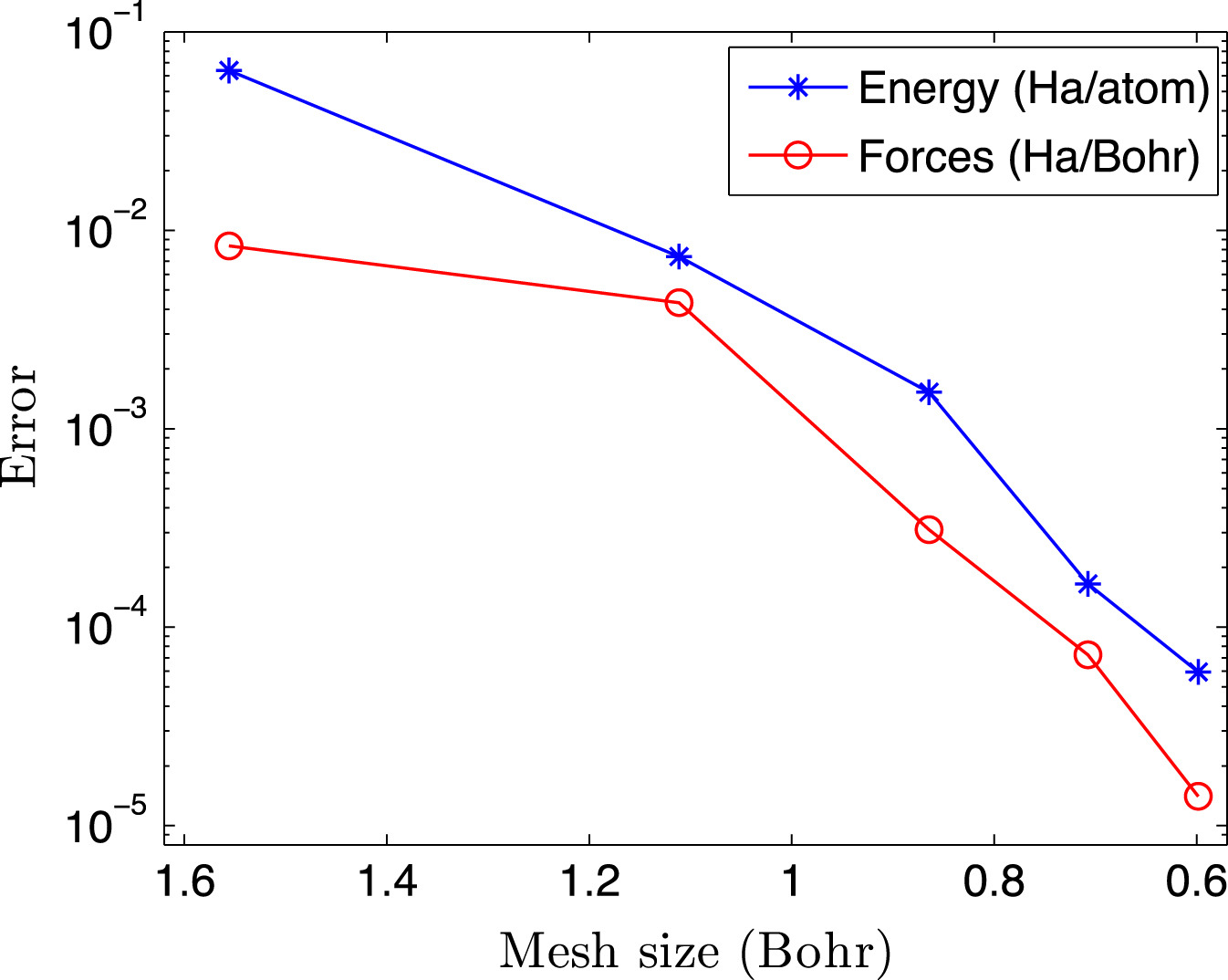
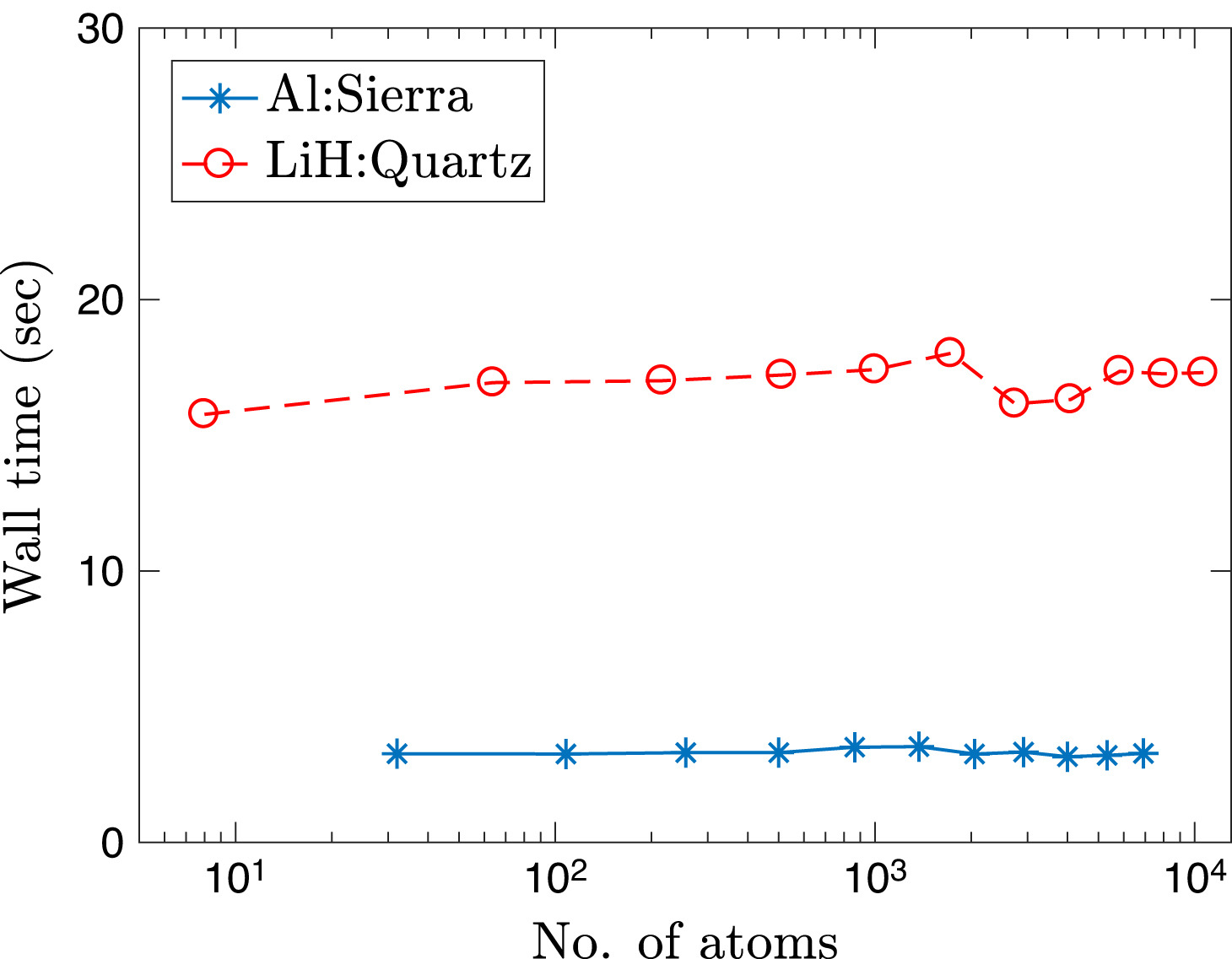
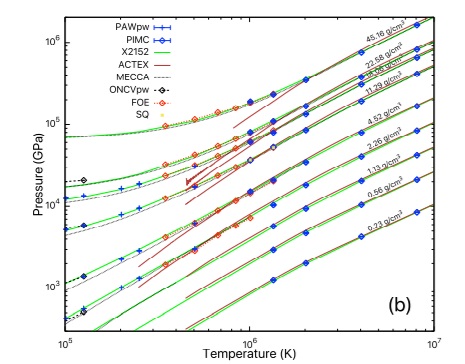
High temperature calculations present additional challenges for DFT. These include the need for a significantly larger number of orbitals to be computed, as the number of partially occupied states increases, and need for more diffuse orbitals, as higher-energy states become less localized. Consequently, cubic-scaling methods as well as local-orbital based linear-scaling methods have very large prefactors, which makes them unsuitable for the study of materials under extreme conditions. In order to overcome these limitations, in the framework provided by the SPARC formulation and implementation, we have recently developed a linear-scaling DFT formulation and implementation referred to as SQDFT [SQDFT1, SQDFT2, SQDFT3], whose cost actually decreases with increasing temperature. Furthermore, it can efficiently scale up to a hundred thousands computational processors, and is therefore able to simulate systems whose sizes are two orders of magnitude larger than previously feasible. SQDFT is currently being utilized to study a variety of materials systems at extreme conditions.


