Ab-initio framework for the study of crystal defects
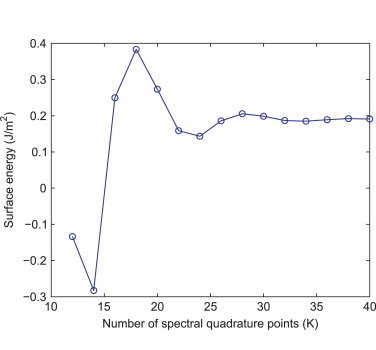
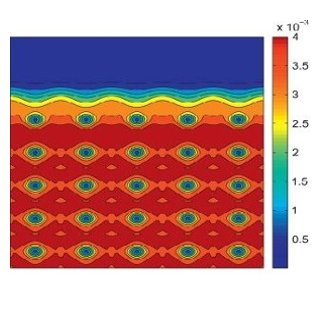
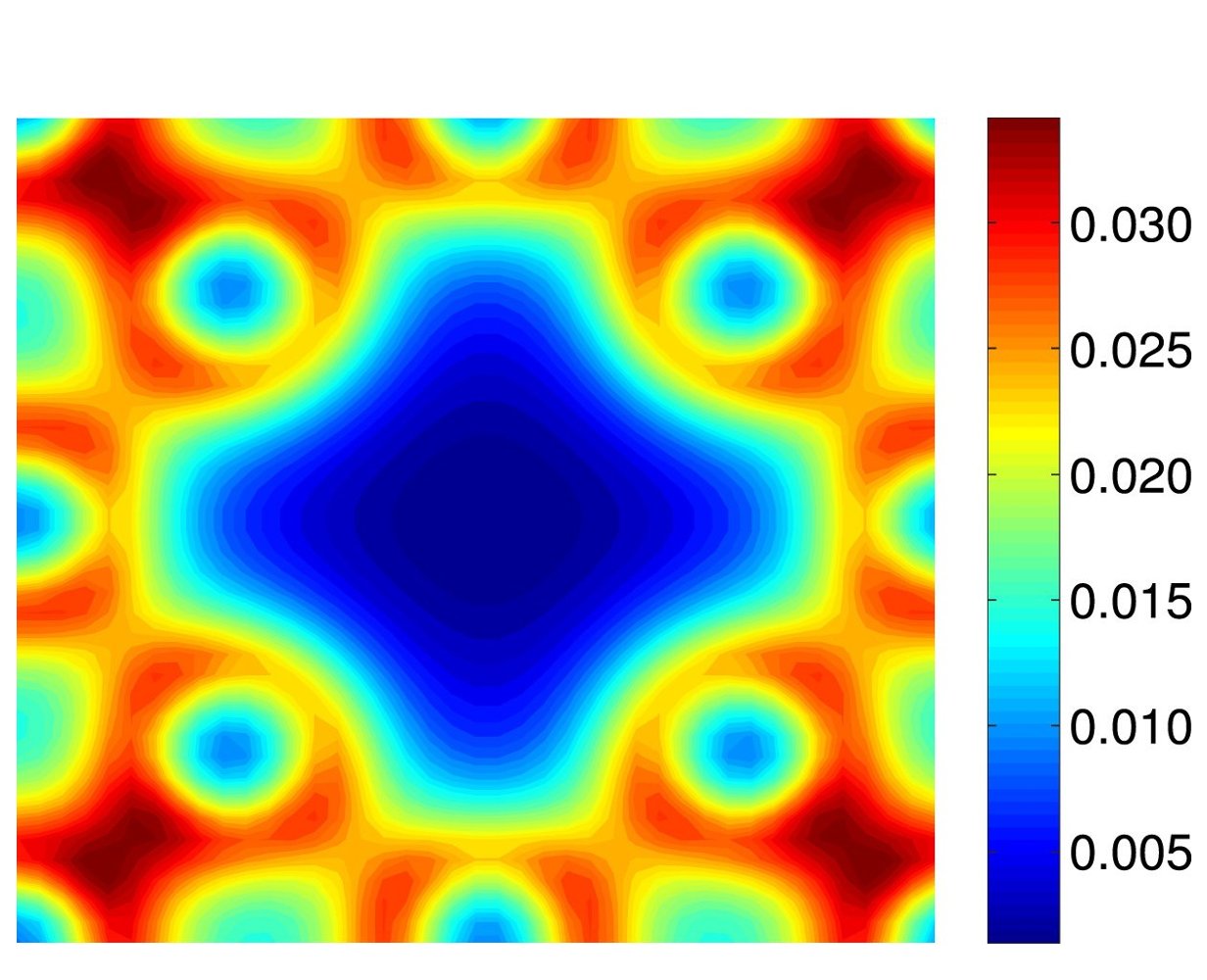
Crystal defects, though present in relatively minute concentrations, play a significant role in determining material properties. This necessitates an accurate characterization of defects at physically relevant defect concentrations (parts per million), which represents a unique challenge since both the electronic structure of the defect core as well as the long range elastic field need to be resolved simultaneously. Since routine DFT calculations are limited to hundreds of atoms, this represents a truly challenging open problem. In order to solve this, we have developed a method to coarse-grain DFT that is solely based on approximation theory, without the introduction of any new equations and resultant spurious physics [CGDFT1]. This work has opened an avenue for the study of extended crystal defects using DFT, which represents a vital step towards understanding the deformation and failure mechanisms in solids. We are currently utilizing this framework to characterize dislocations, the interactions between them, and their interaction with macroscopic fields (e.g. strain).


