Publications
BOOK CHAPTER
.png)
Kohn-Sham DFT: Coarse-graining
Accurate approximations of density functional theory for large systems with applications to defects in crystalline solids
JOURNAL
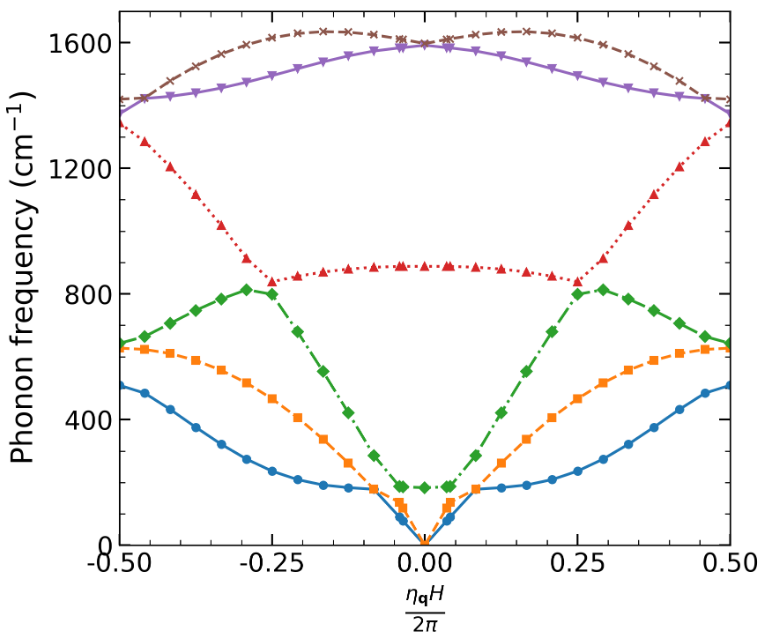
Kohn-Sham DFT: Cyclix phonons
Cyclic- and helical-symmetry-adapted phonon formalism within density functional perturbation theory
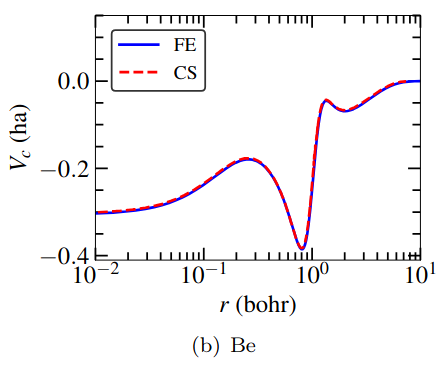
Kohn-Sham DFT: RPA-OEP Atomic Structure
Spectral finite-element formulation of the optimized effective potential method for atomic structure in the random phase approximation
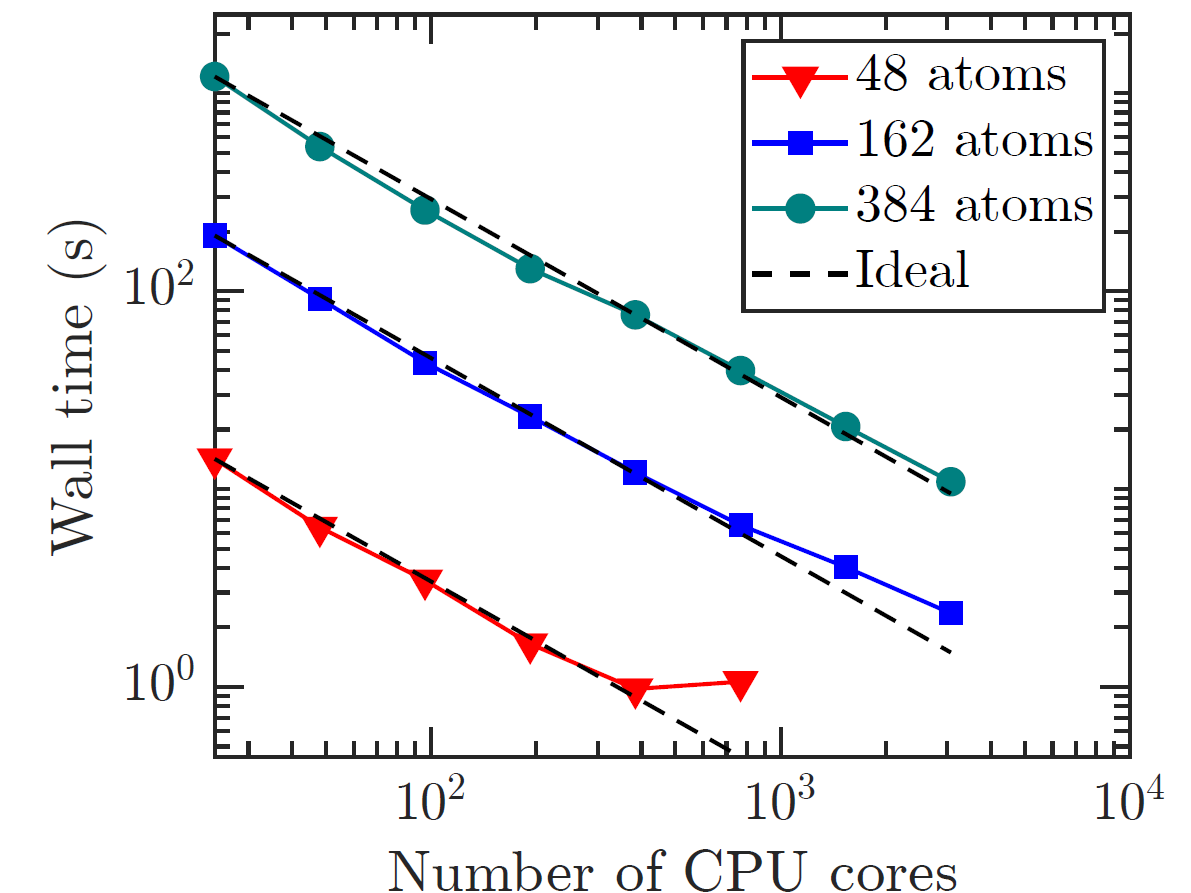

Vacancy clustering in aluminum
Ab initio study of strain-driven vacancy clustering in aluminum
Phys. Rev. B (2025)
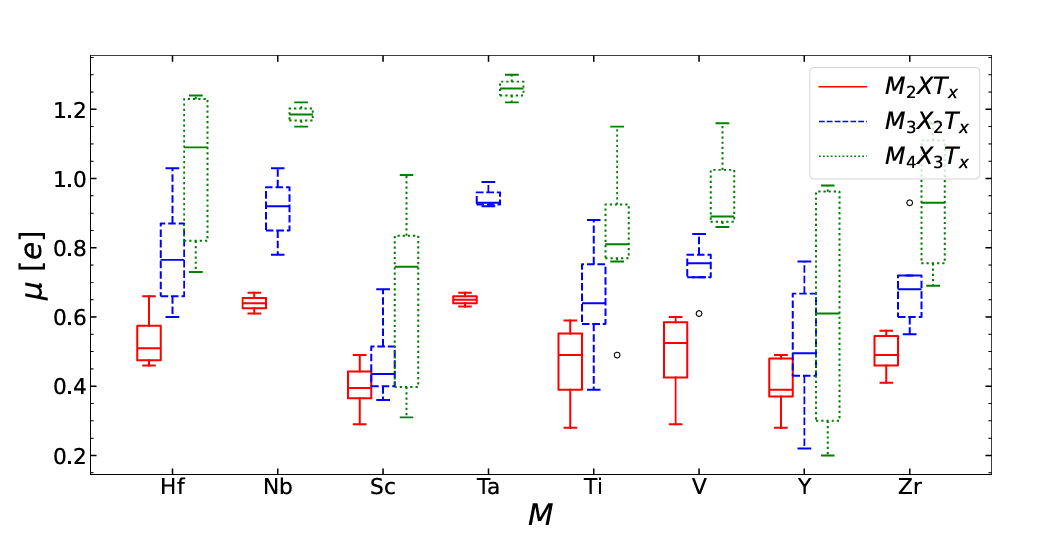
MXene monolayers: Flexoelectric effect
Ab initio study of flexoelectricity in MXene monolayers
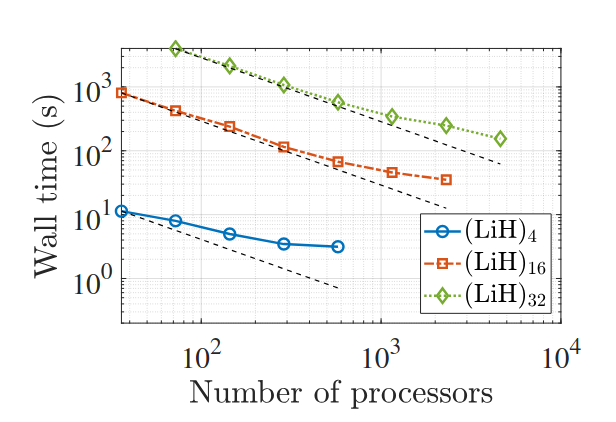
Kohn-Sham DFT: DFPT-RPA correlation energy
Random Phase Approximation Correlation Energy using Real-Space Density Functional Perturbation Theory
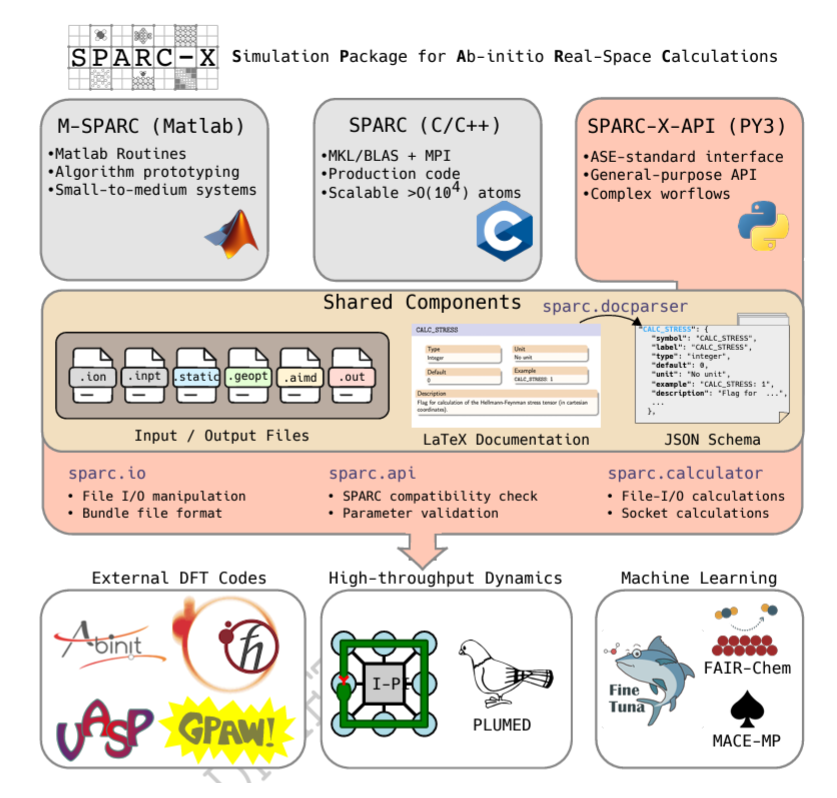
Kohn-Sham DFT: SPARC-X API
SPARC-X-API: Versatile Python Interface for Real-space Density Functional Theory Calculations
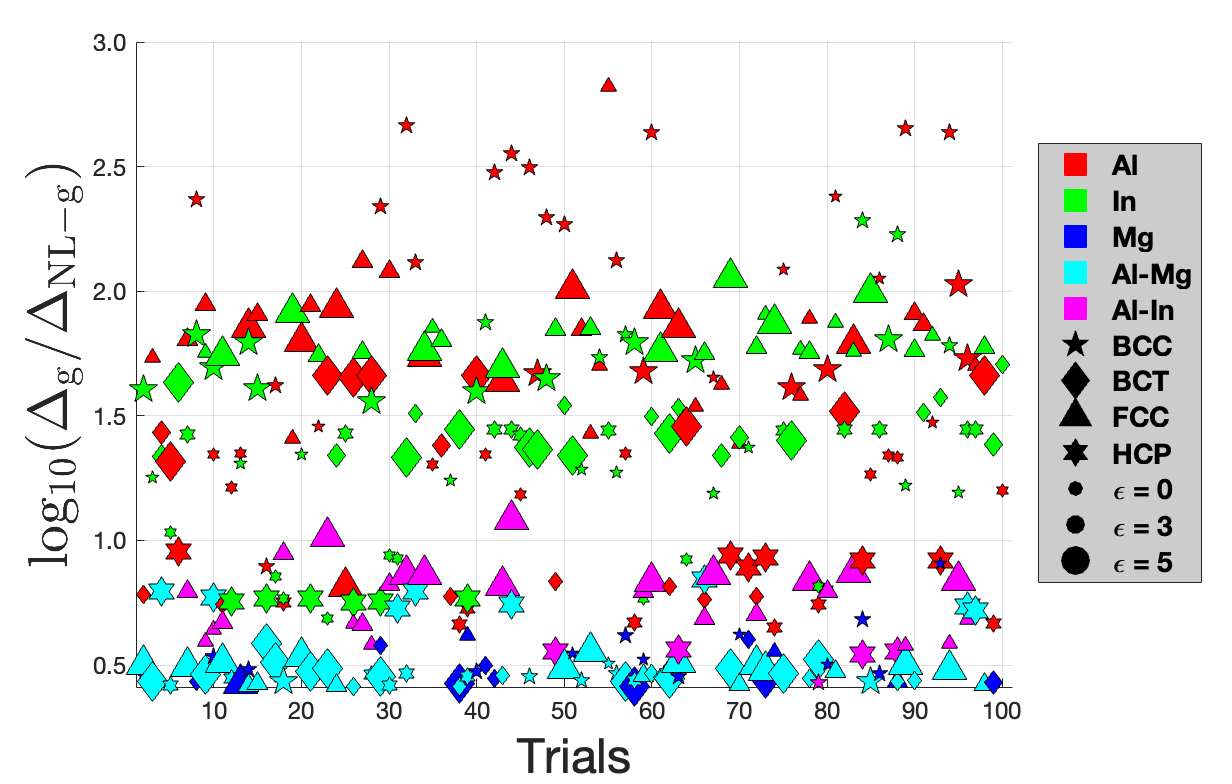
Orbital-free DFT: Kinetic energy density functional
Orbital-free density functionals based on real and reciprocal space separation
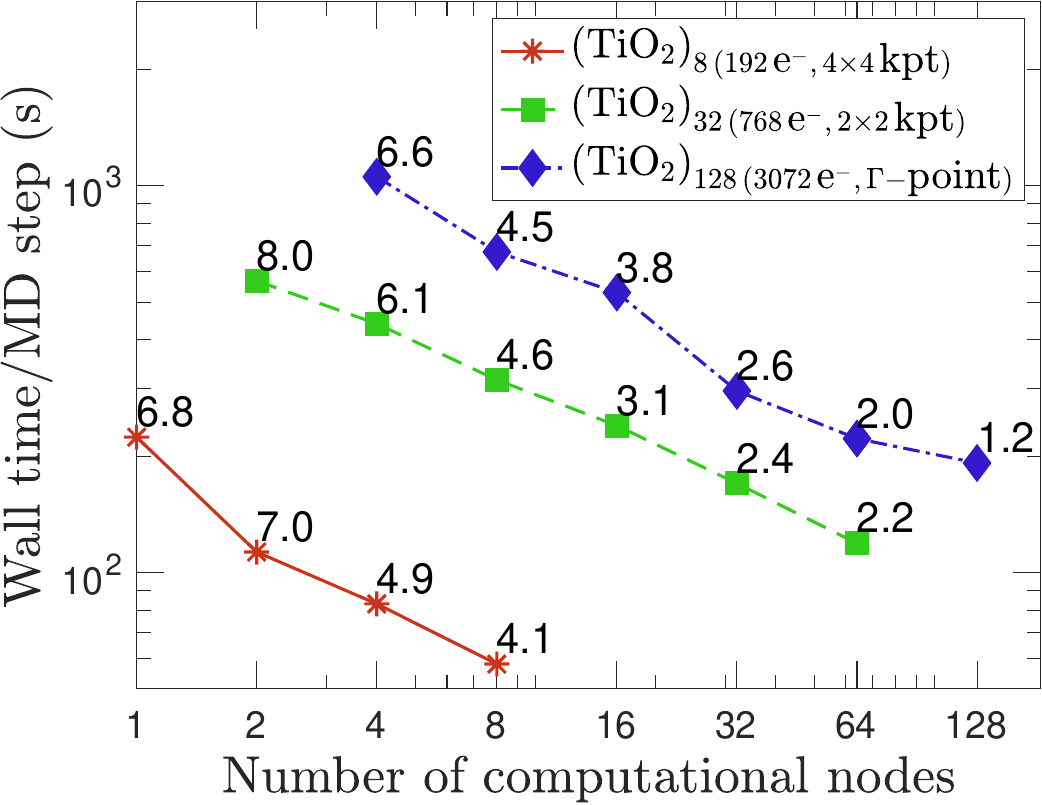
Kohn-Sham DFT: SPARC GPU acceleration of hybrid XC
GPU acceleration of hybrid functional calculations in the SPARC electronic structure code
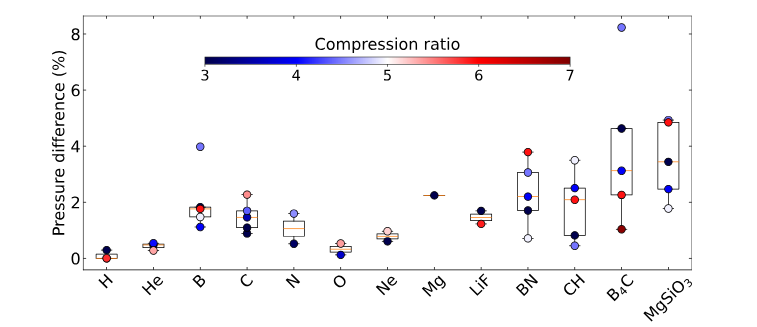
Kohn-Sham DFT: Accuracy for WDM and HDM
Accuracy of Kohn-Sham density functional theory for warm- and hot-dense matter equation of state
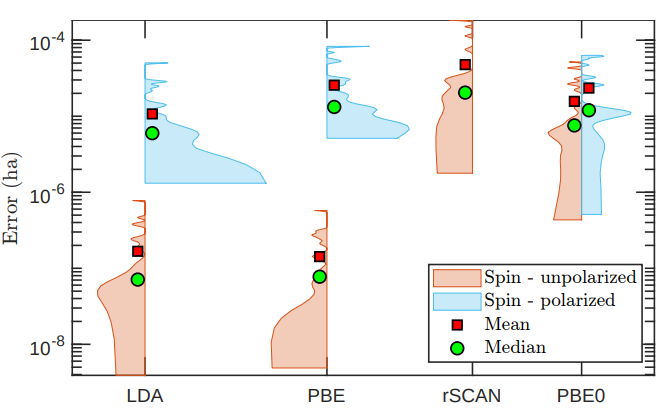
Kohn-Sham DFT: Atomic structure
Spectral scheme for atomic structure calculations in density functional theory
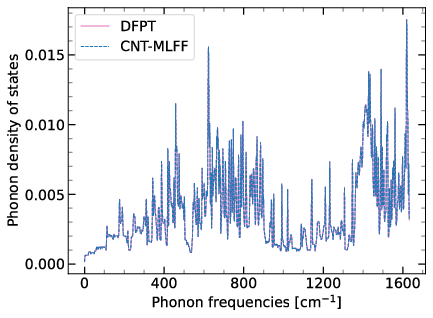
Cyclix MLFF: Phonon
Cyclic and helical symmetry-informed machine learned force fields: Application to lattice vibrations in carbon nanotubes
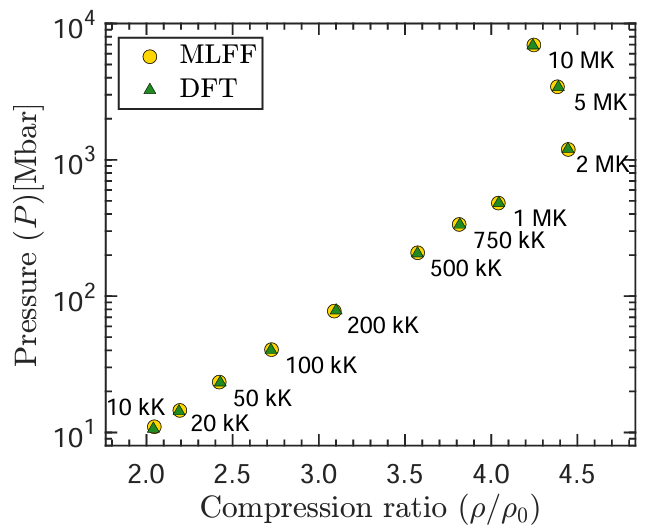
On-the-fly MLFF: Shock Hugoniot
Shock Hugoniot calculations using on-the-fly machine learned force fields with ab initio accuracy
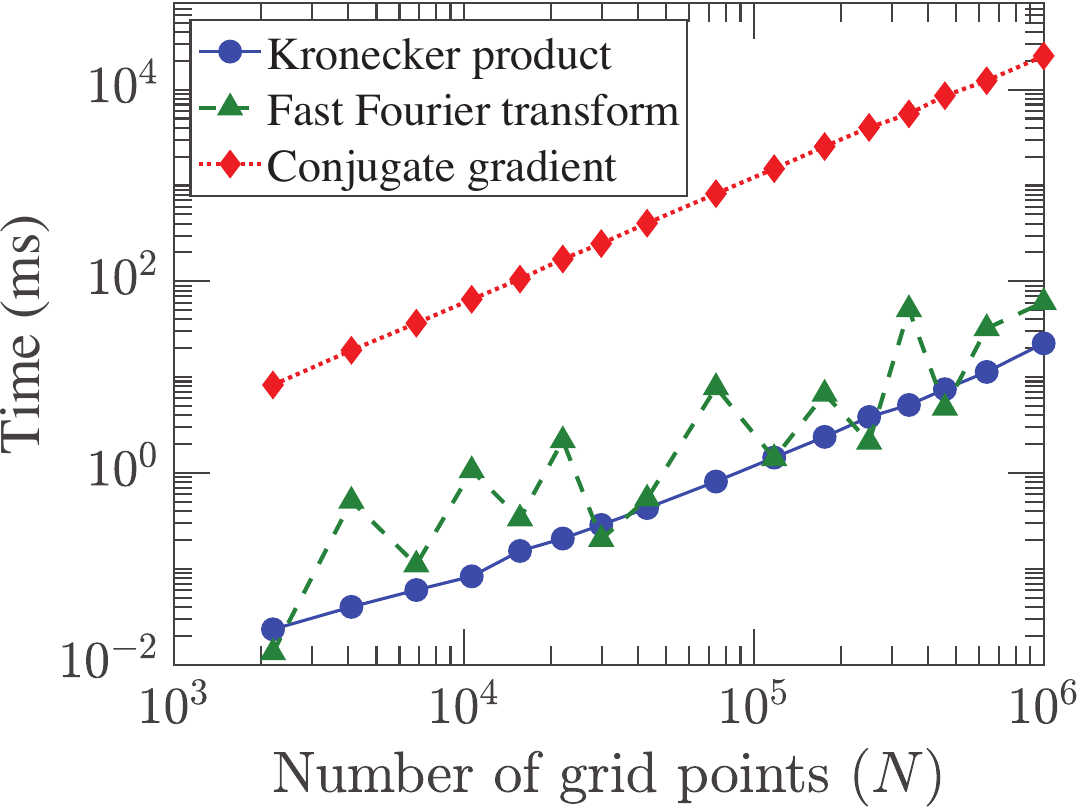
Kohn-Sham DFT: Hybrid functionals
Efficient real space formalism for hybrid density functionals
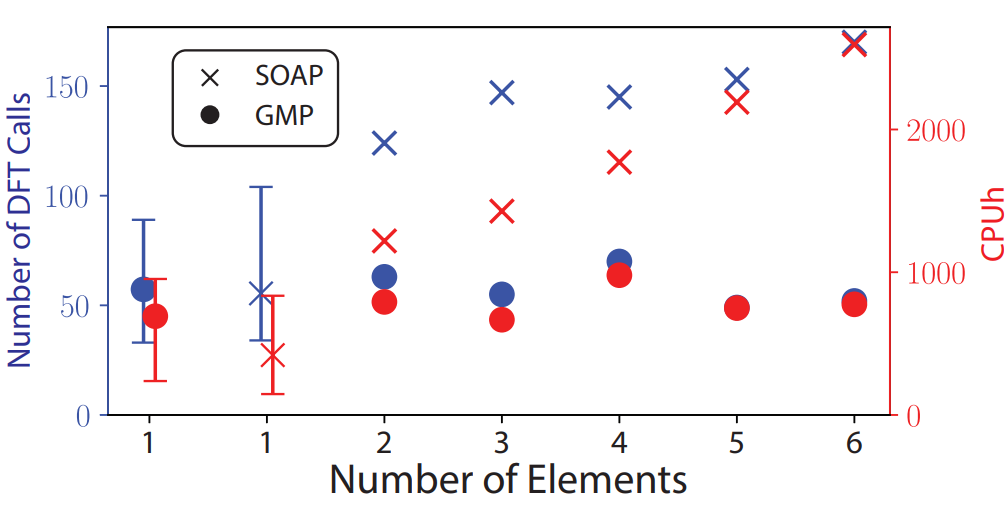
On-the-fly MLFF: GMP Descriptor
Overcoming the chemical complexity bottleneck in on-the-fly machine learned molecular dynamics simulations
Review: TCCW2
Review of the second charged-particle transport coefficient code comparison workshop
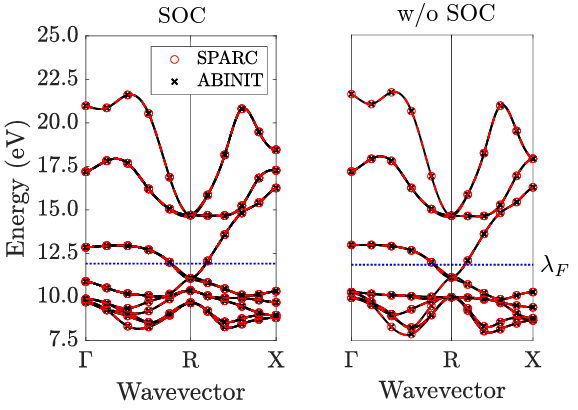
Kohn-Sham DFT: SPARC software v2.0.0
SPARC v2.0.0: Spin-orbit coupling, dispersion interactions, and advanced exchange-correlation functionals
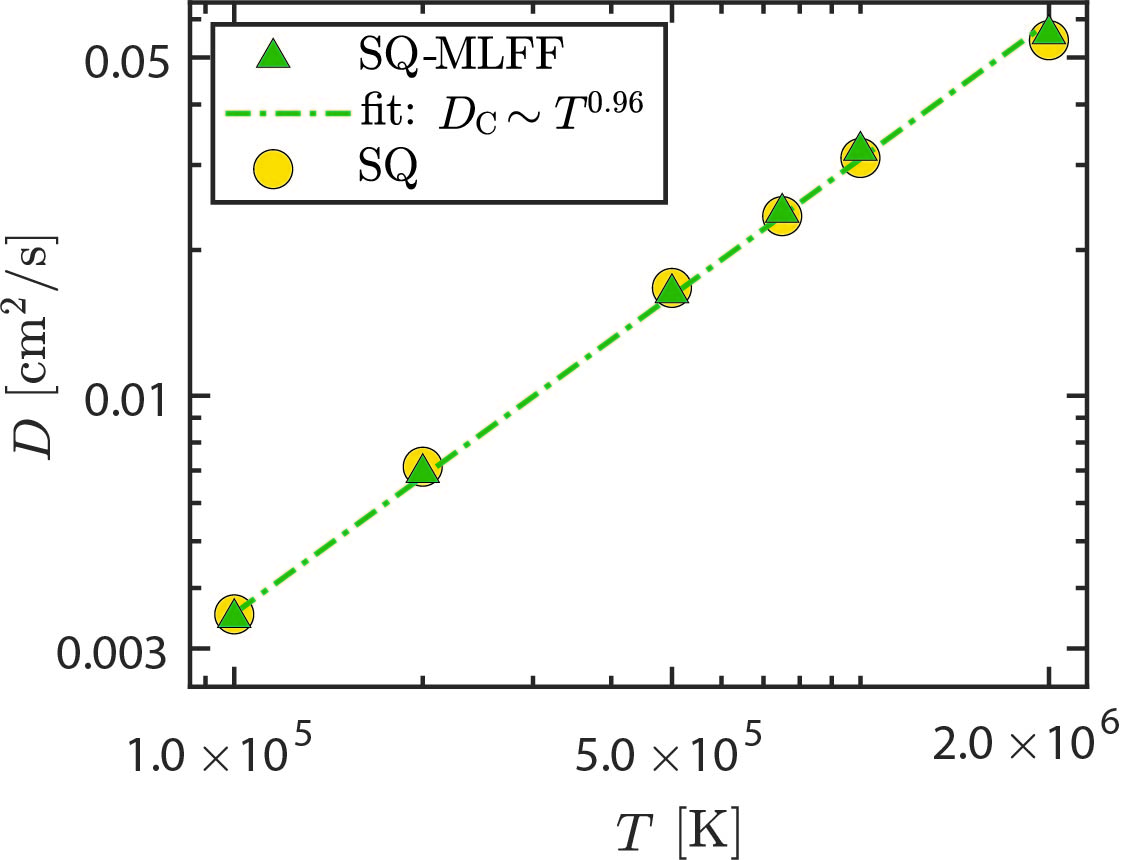
On-the-fly MLFF: WDM
On-the-fly machine learned force fields for the study of warm dense matter: Application to diffusion and viscosity of CH

Kohn-Sham DFT: Machine learning XC
Self-consistent convolutional density functional approximations: Application to adsorption at metal surfaces
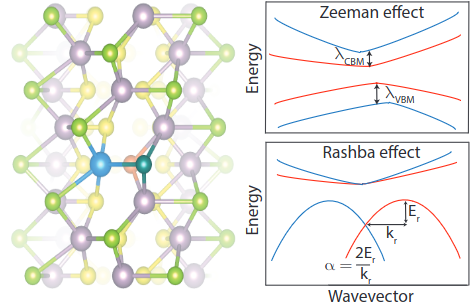
TMD and Janus TMD nanotubes: Zeeman and Rashba effects
Strain engineering of Zeeman and Rashba effects in transition metal dichalcogenide nanotubes and their Janus variants: An ab initio study
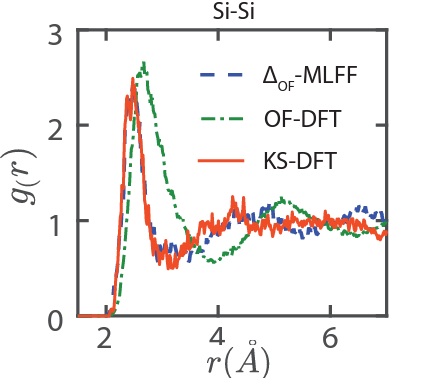
Orbital free to Kohn-Sham DFT: Δ-Machine learning
Kohn-Sham accuracy from orbital-free density functional theory via Δ-machine learning
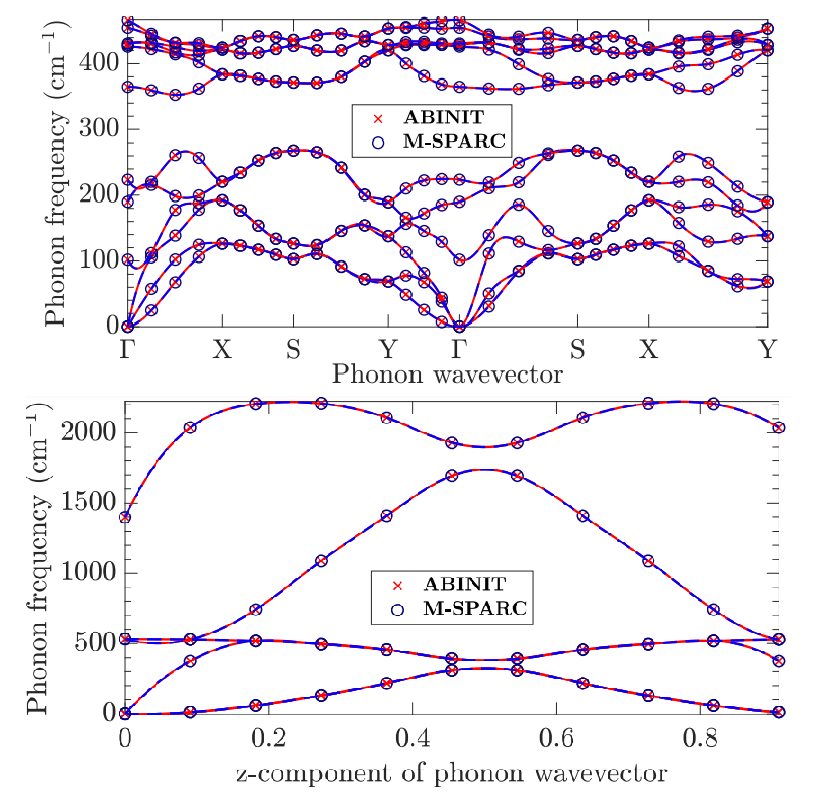
Kohn-Sham DFT: Phonons
Calculation of phonons in real-space density functional theory
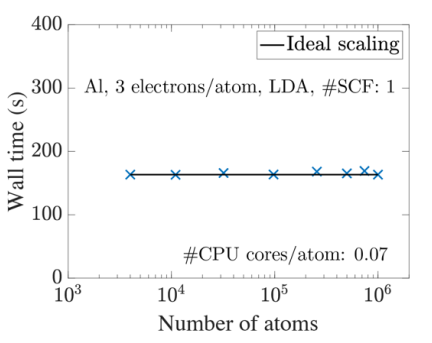
Kohn-Sham DFT: SPARC software
Roadmap on Electronic Structure Codes in the Exascale Era
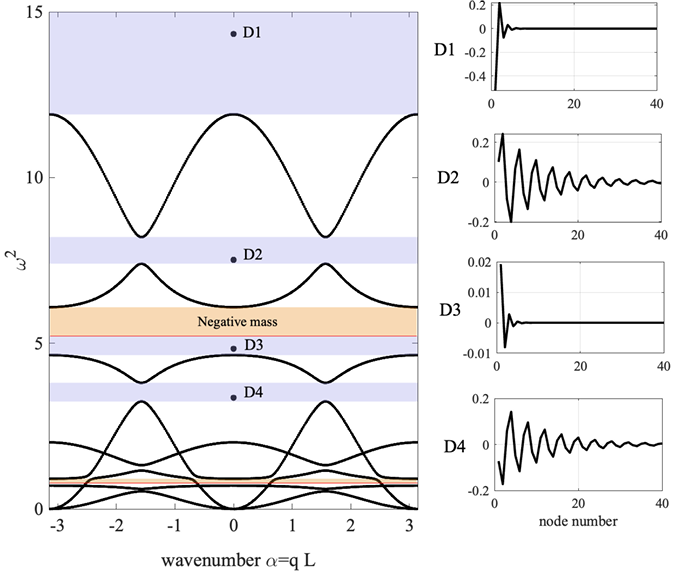
Effective mass: lattice with microstructure
On the effective dynamic mass of mechanical lattices with microstructure
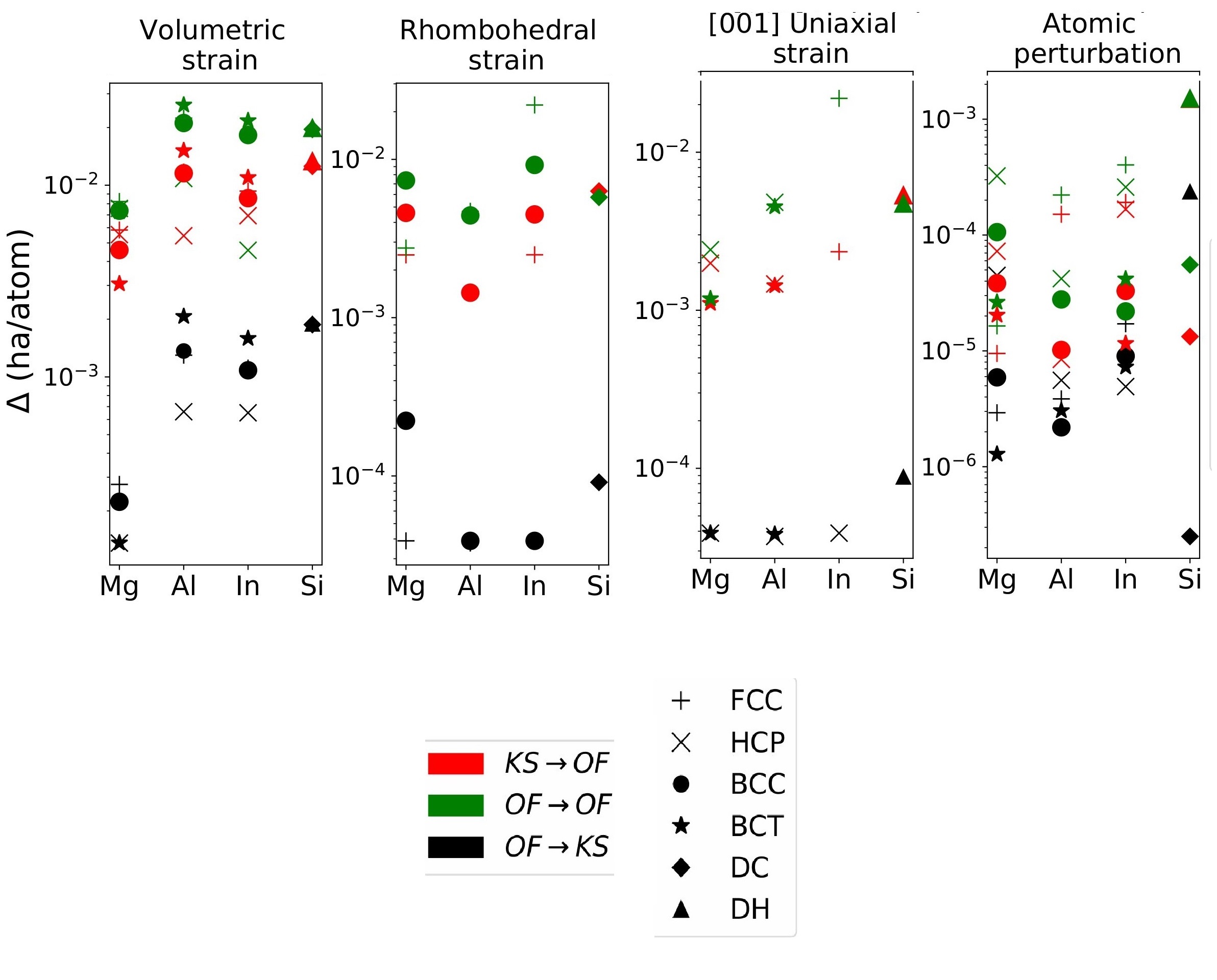
Orbital-free DFT: Error
Assessing the source of error in the Thomas–Fermi–von Weizsäcker density functional
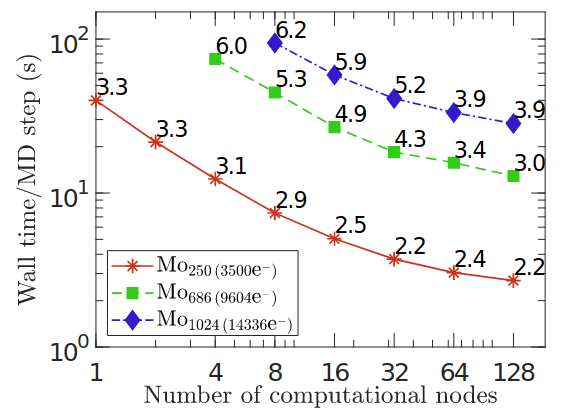
Kohn-Sham DFT: SPARC GPU acceleration of local XC
GPU acceleration of local and semilocal density functional calculations in the SPARC electronic structure code
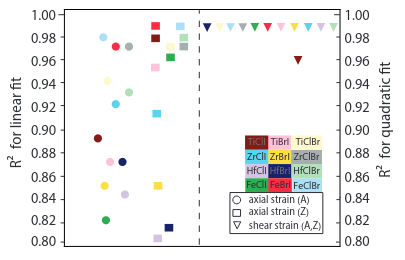
Janus TMH nanotubes: Strain engineering
Ab initio study on the electromechanical response of Janus transition metal dihalide nanotubes
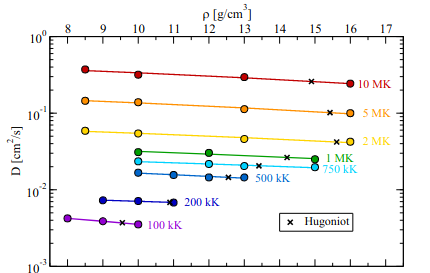
Properties of warm and hot dense carbon
Properties of carbon up to 10 million kelvin from Kohn-Sham density functional theory molecular dynamics
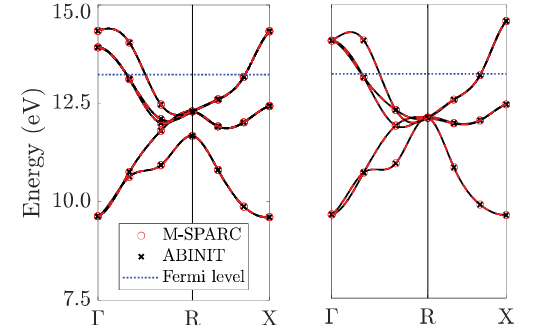
Kohn-Sham DFT: M-SPARC software v2.0.0
Version 2.0.0 -- M-SPARC: Matlab-Simulation Package for Ab-initio Real-space Calculations
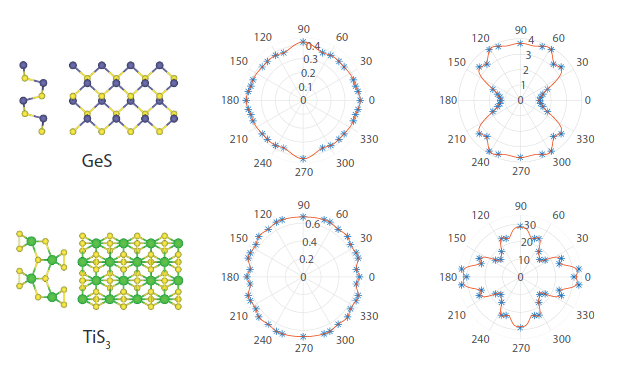
Rectangular monolayers: Bending along different directions
On the bending of rectangular atomic monolayers along different directions: an ab initio study
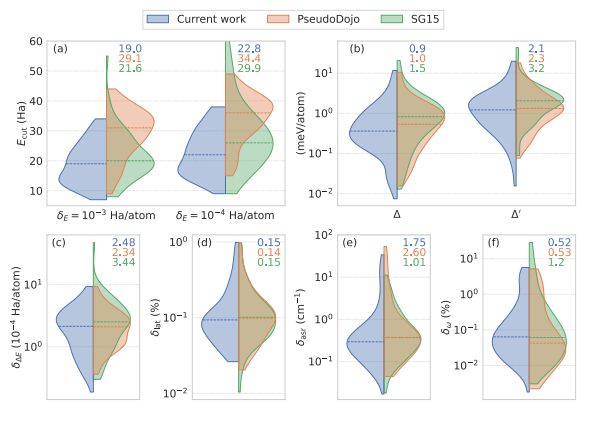
Kohn-Sham DFT: Table of Pseudopotentials
Soft and transferable pseudopotentials from multi-objective optimization
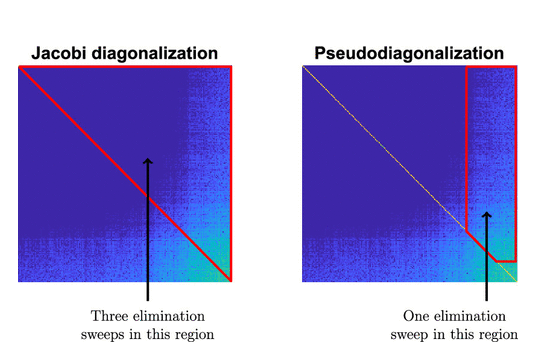
Kohn-Sham DFT: Pseudodiagonalization
Pseudodiagonalization Method for Accelerating Nonlinear Subspace Diagonalization in Density Functional Theory
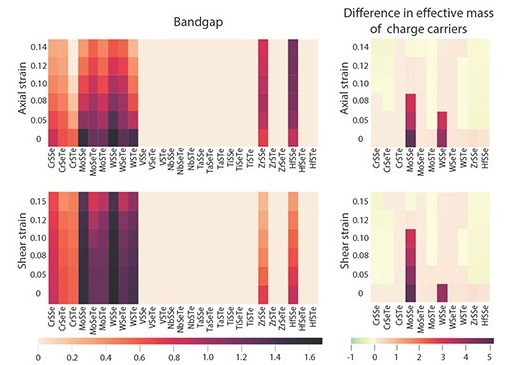
Janus TMD nanotubes: Strain engineering
Strain engineering of Janus transition metal dichalcogenide nanotubes: An ab intio study
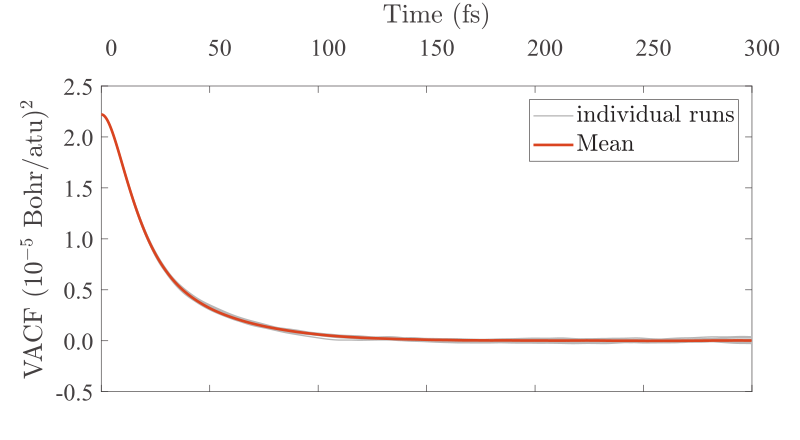
Kohn-Sham DFT: SQ3 method
Real-space density kernel method for Kohn-Sham density functional theory calculations at high temperature
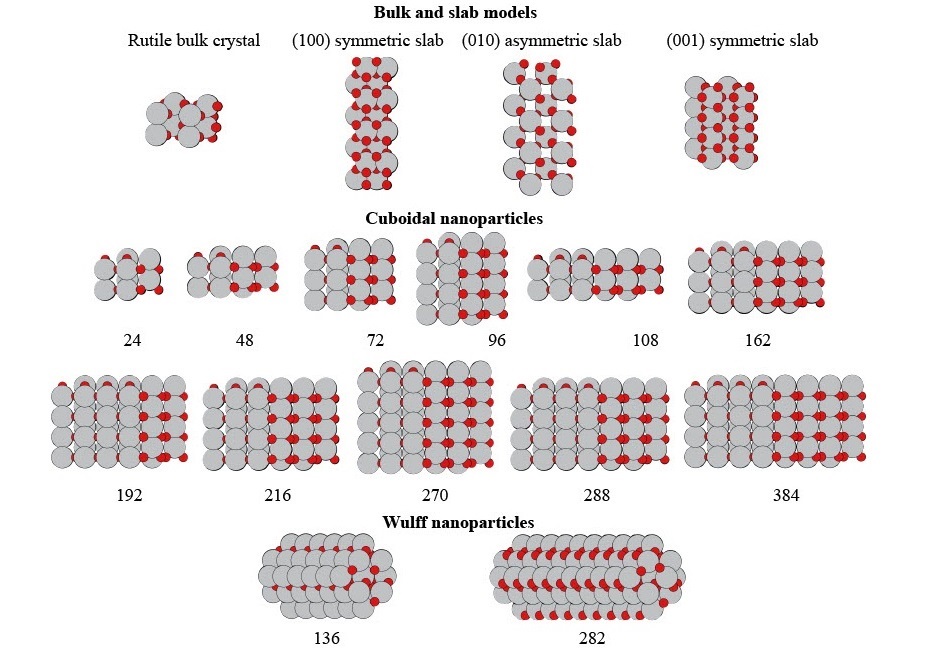
Rutile TiO2: Finite-size effects
Ab-initio investigation of finite size effects in rutile titania nanoparticles with semilocal and nonlocal density functionals
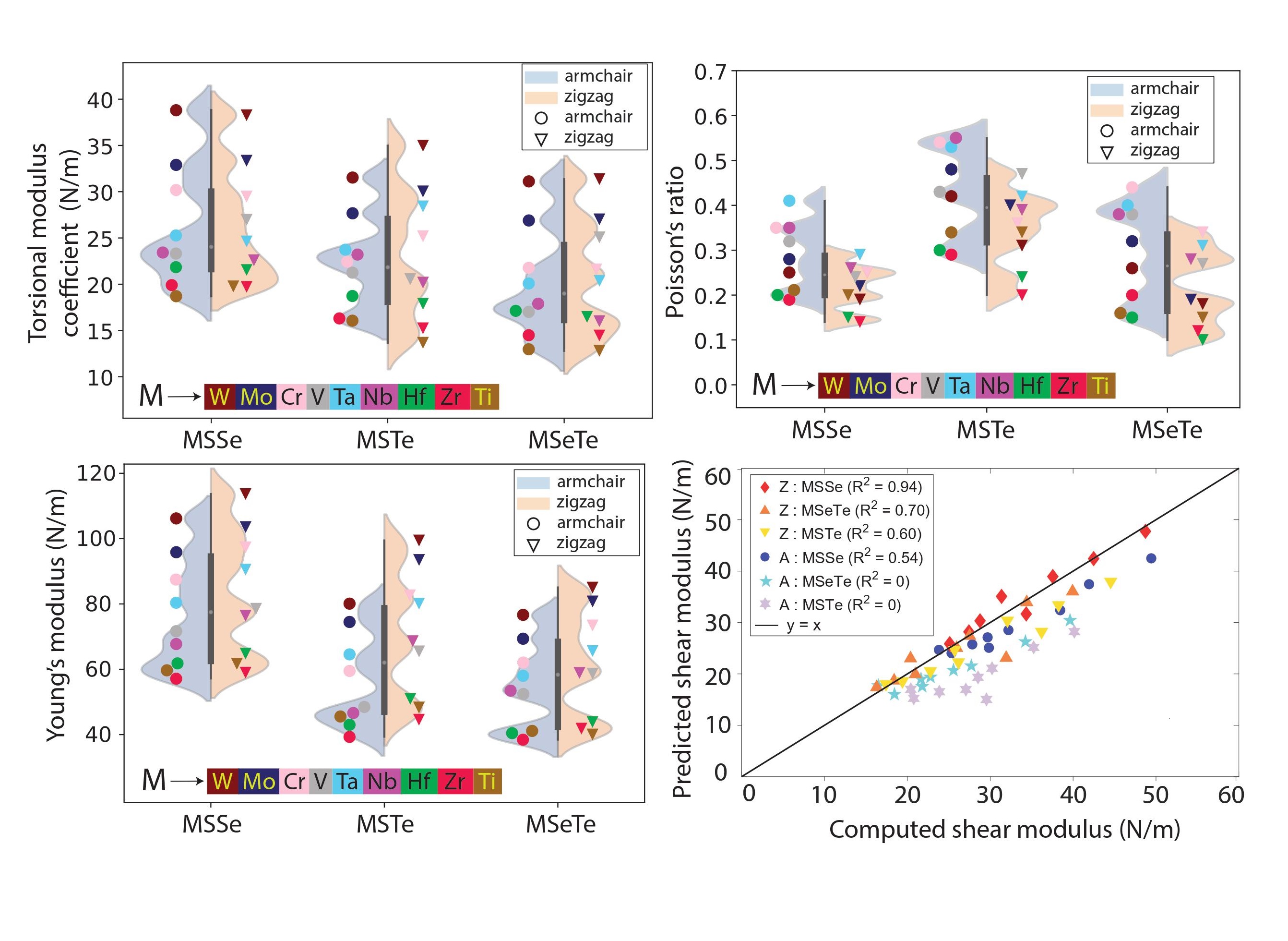
Janus TMD nanotubes: Elastic properties
Elastic properties of Janus transition metal dichalcogenide nanotubes from first principles
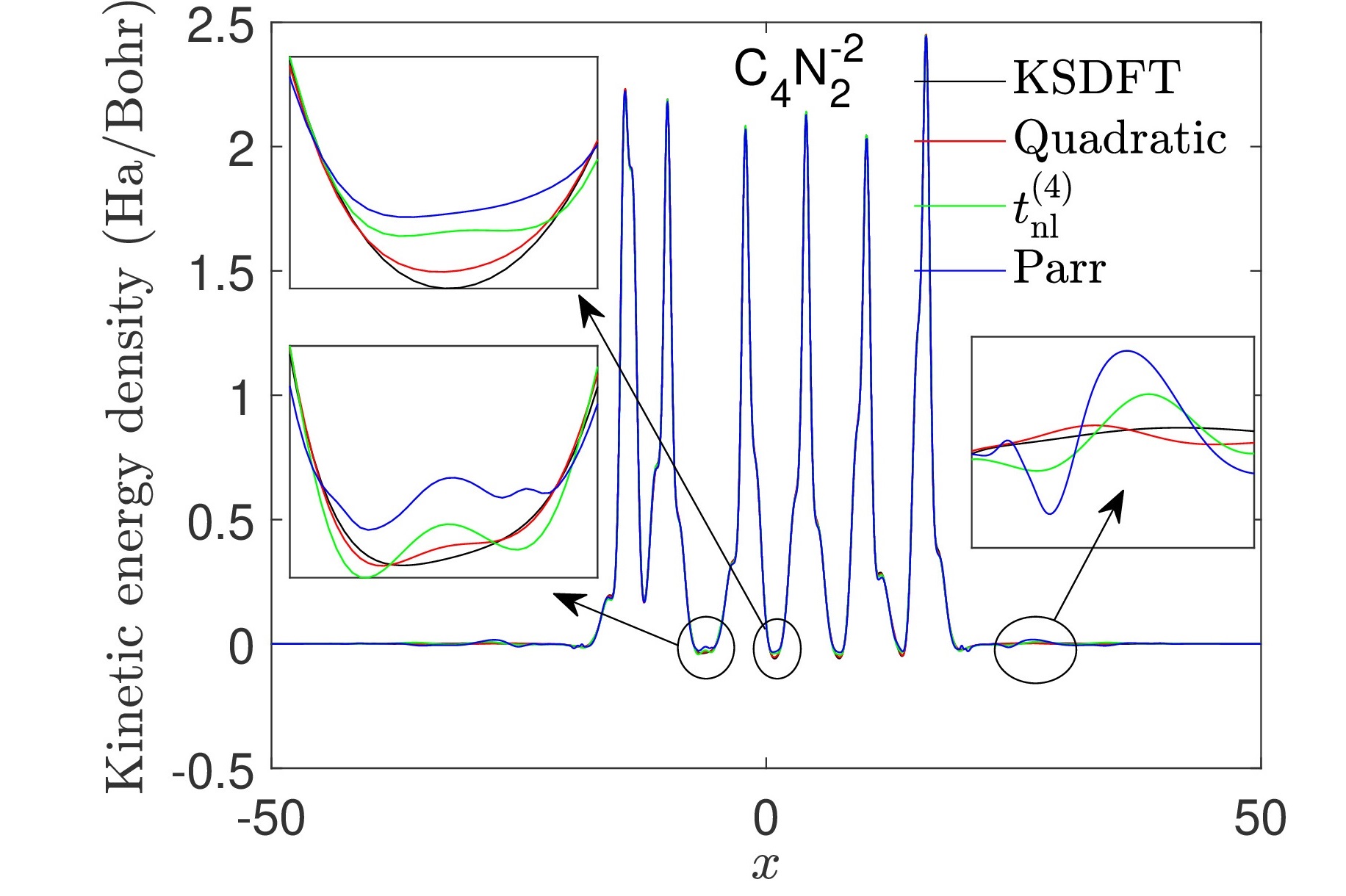
1D Kohn-Sham model: Exact exchange surrogate model
Accurate parametrization of the kinetic energy functional for calculations using exact exchange
TMD nanotubes: Strain engineering
Torsional strain engineering of transition metal dichalcogenide nanotubes: An ab initio study
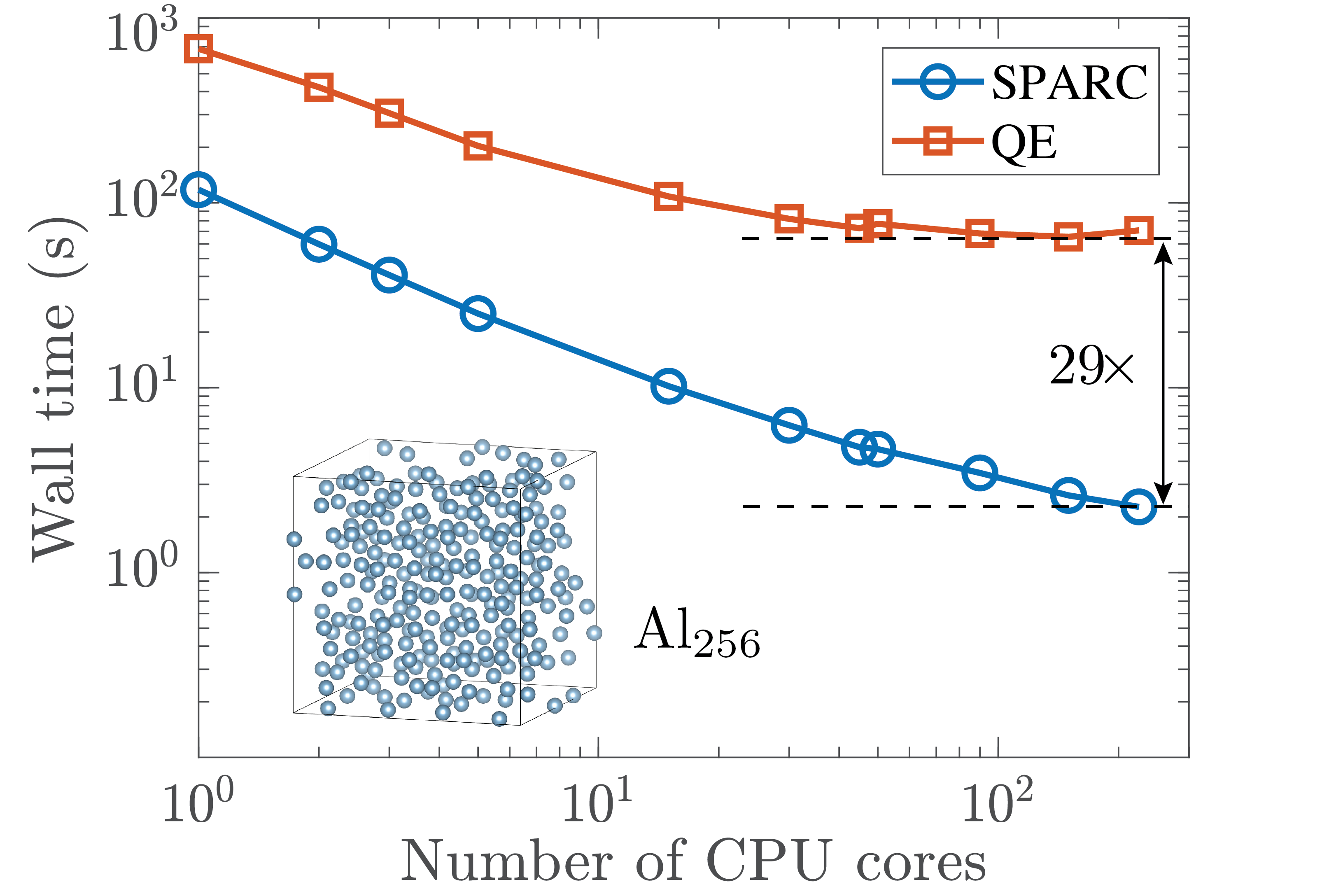
Kohn-Sham DFT: SPARC software
SPARC: Simulation Package for Ab-initio Real-space Calculations
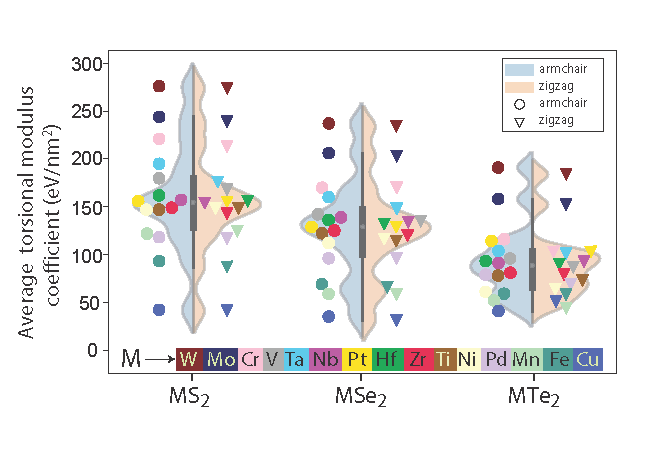
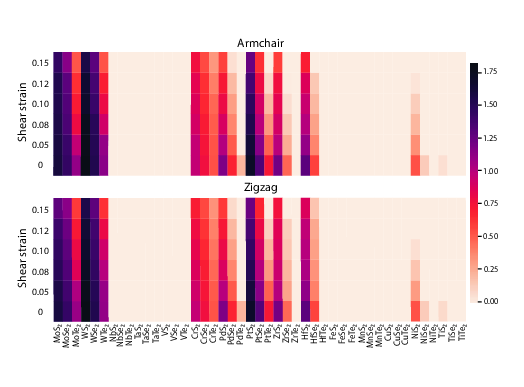
TMD nanotubes: Torsional moduli
Torsional moduli of transition metal dichalcogenide nanotubes from first principles
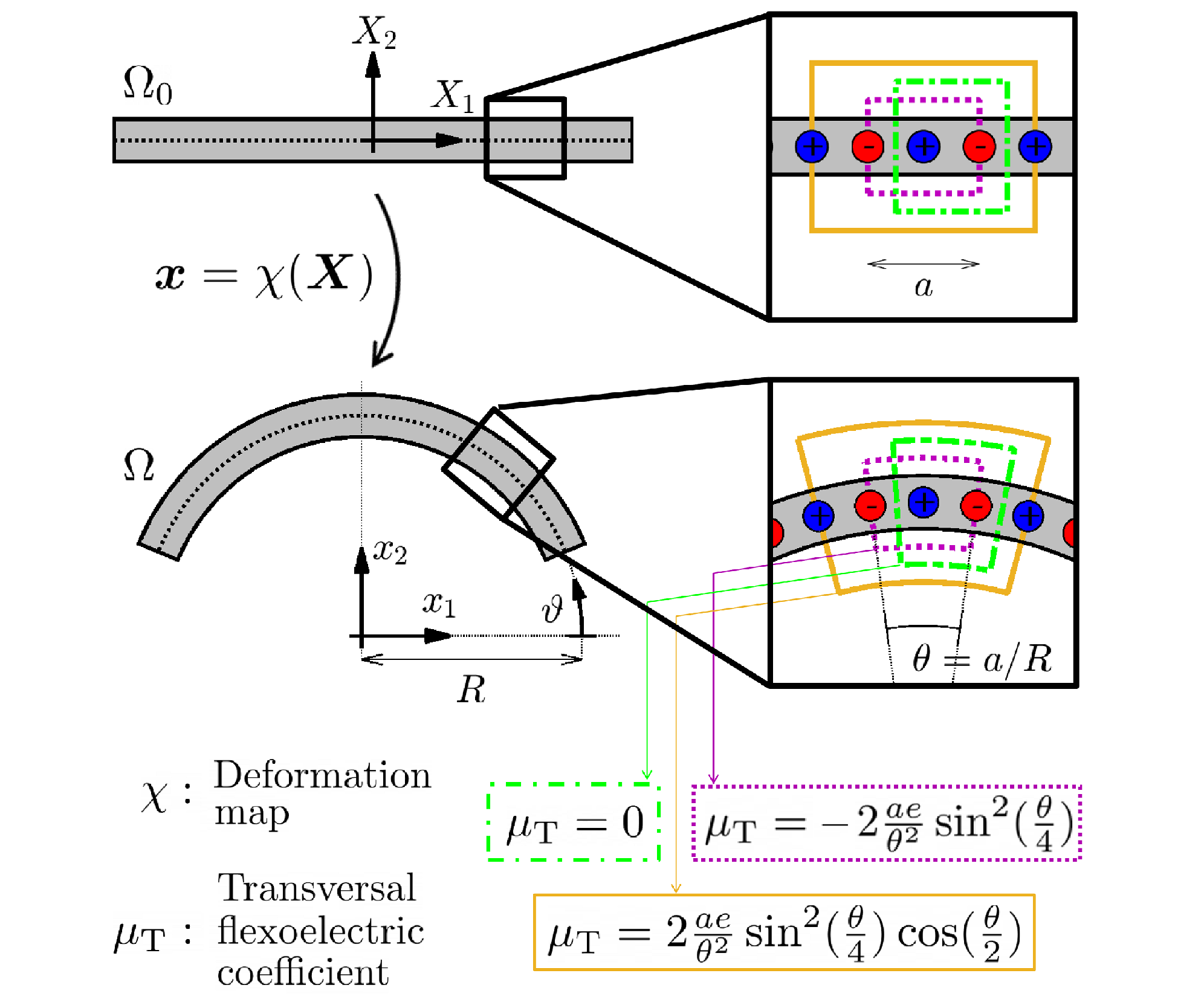
Transversal flexoelectric coefficient: Formulation
Transversal flexoelectric coefficient for nanostructures at finite deformations from first principles
Kohn-Sham DFT: Real-space FLOSIC
Implementation of Perdew–Zunger self-interaction correction in real space using Fermi–Löwdin orbitals
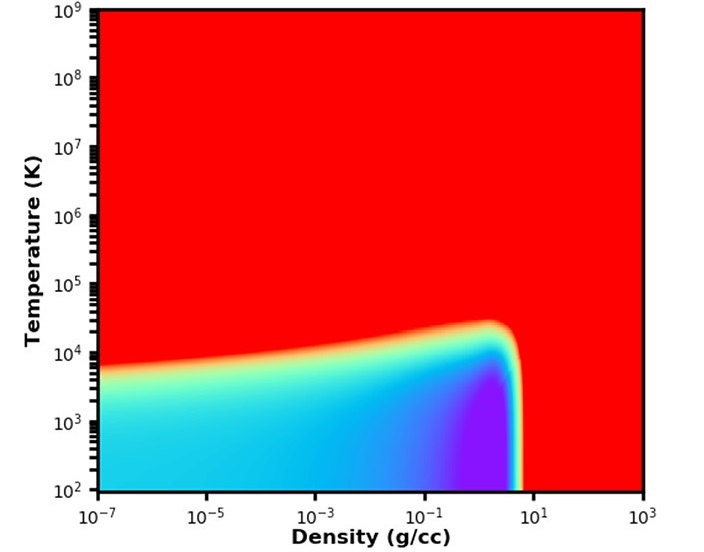
Equation of State: Beryllium
Development of a Multiphase Beryllium Equation of State and Physics-based Variations
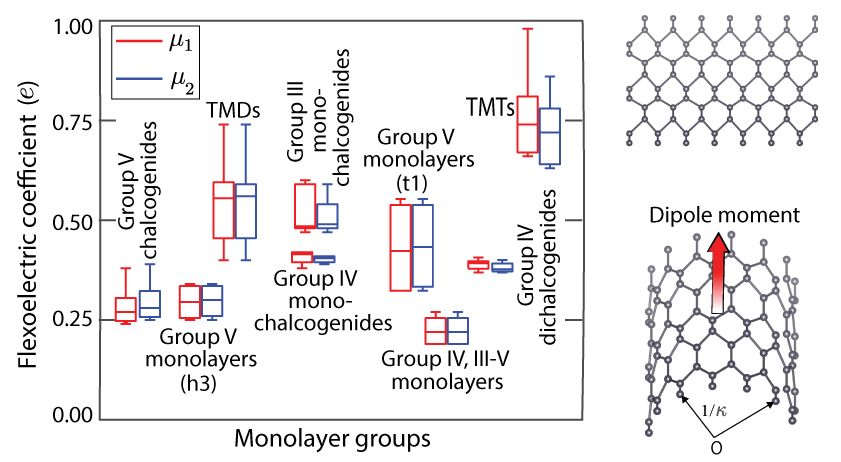
Atomic monolayers: Flexoelectric effect
Flexoelectricity in atomic monolayers from first principles
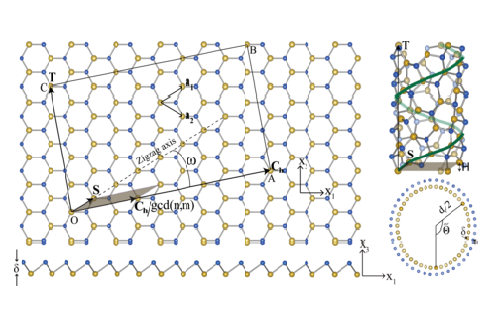
Kohn-Sham DFT: Cyclic+Helical DFT
Real-space density functional theory adapted to cyclic and helical symmetry: Application to torsional deformation of carbon nanotubes
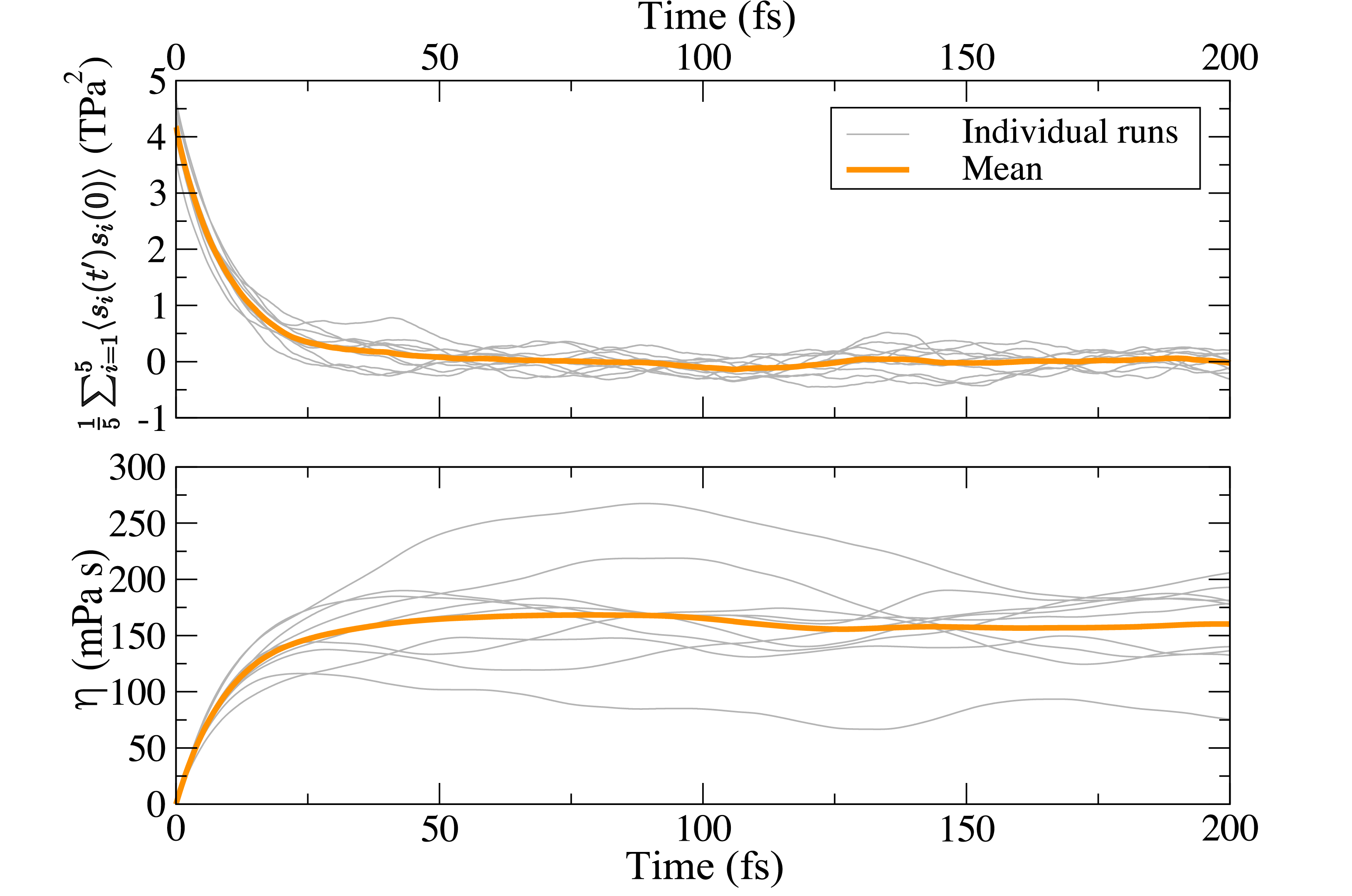
Kohn-Shan DFT: O(N) stress tensor
Real-space formulation of the stress tensor for O(N) density functional theory: application to high temperature calculations
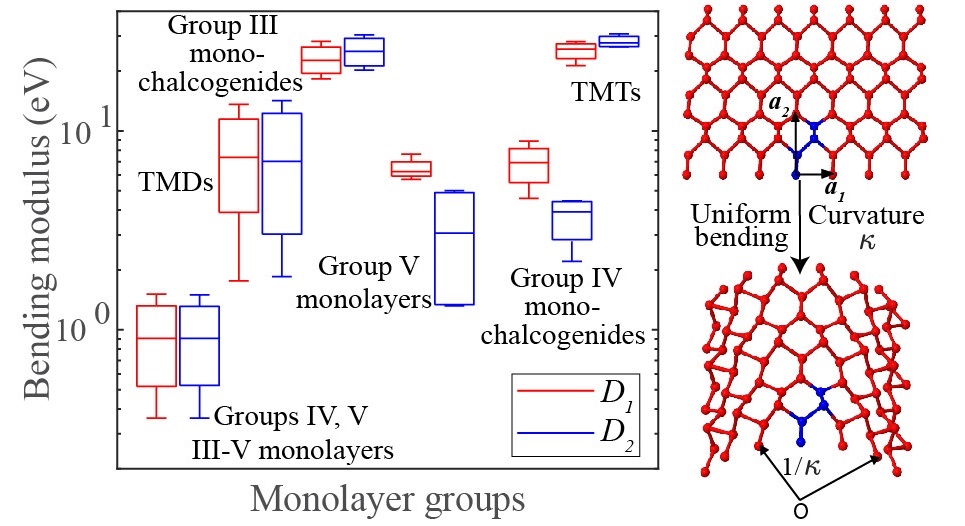
Atomic monolayers: Bending moduli
Bending moduli for forty-four select atomic monolayers from first principles
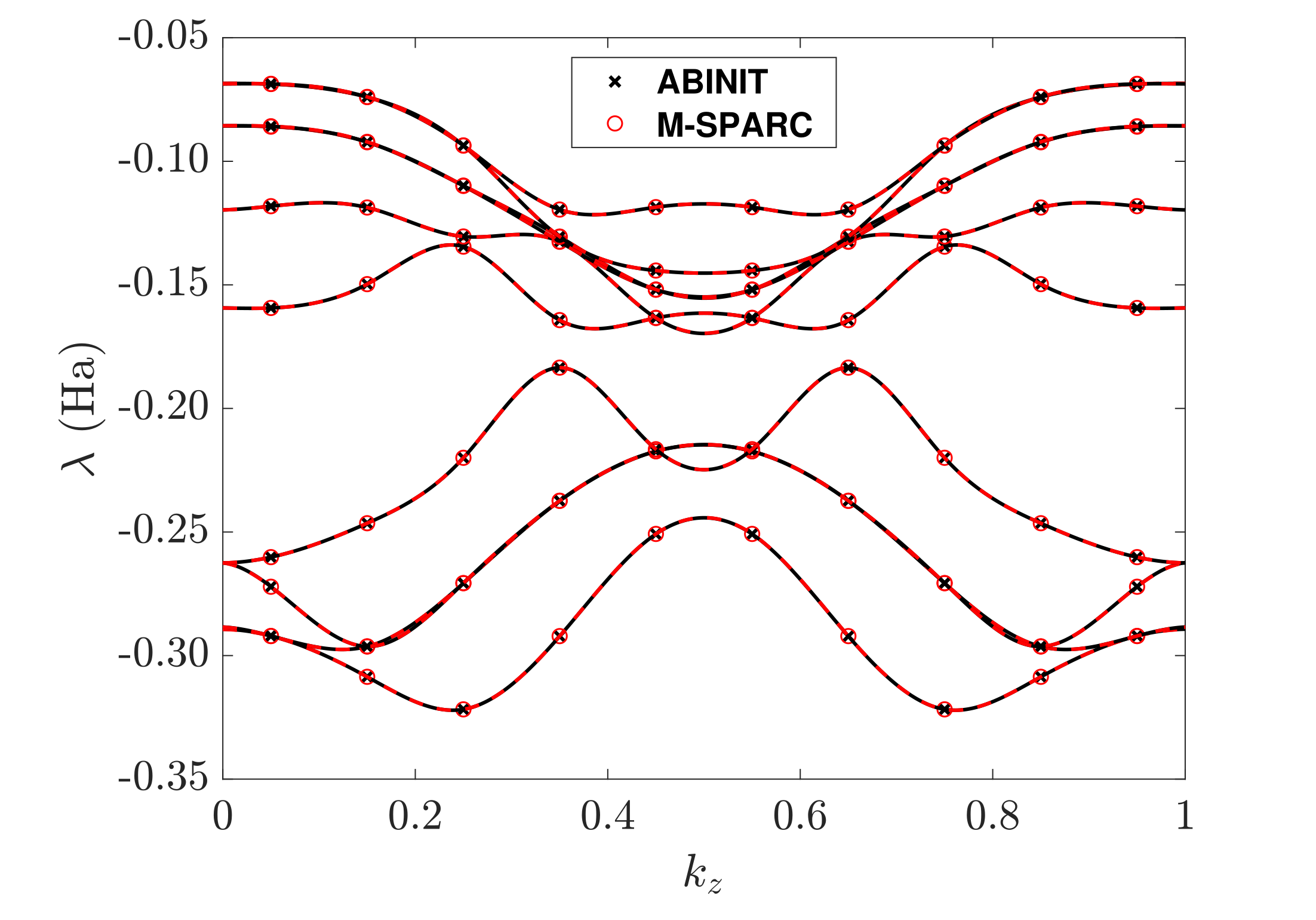
Kohn-Sham DFT: M-SPARC software
M-SPARC: Matlab-Simulation Package for Ab-initio Real-space Calculations

Topology Optimization: Accelerated fixed-point formulation
Accelerated fixed-point formulation of Topology Optimization: application to compliance minimization problems
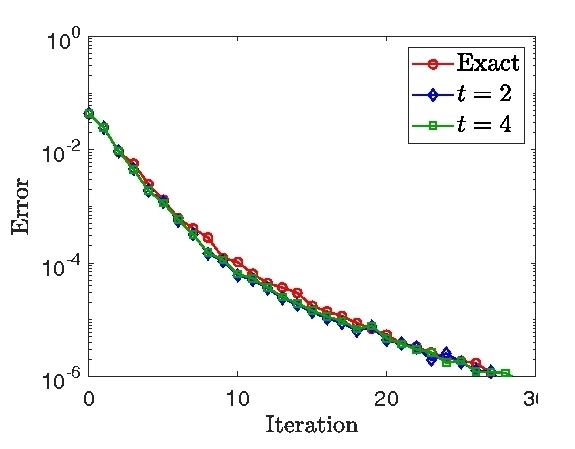
Kohn-Sham DFT: Preconditioning mixing
On preconditioning the self-consistent field iteration in real-space Density Functional Theory
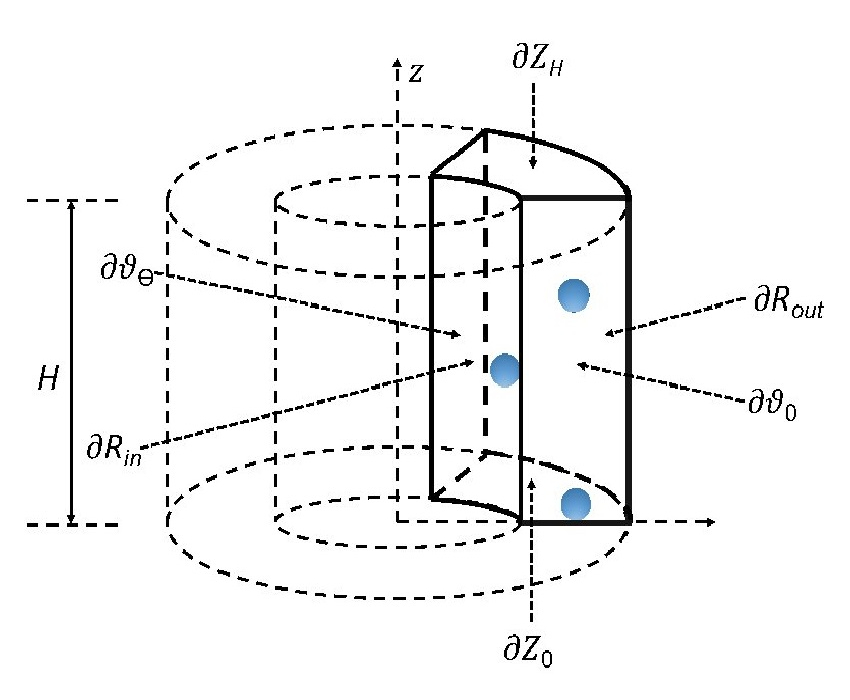
Kohn-Sham DFT: Cyclic DFT II
Symmetry-adapted real-space density functional theory for cylindrical geometries: Application to large group IV nanotubes
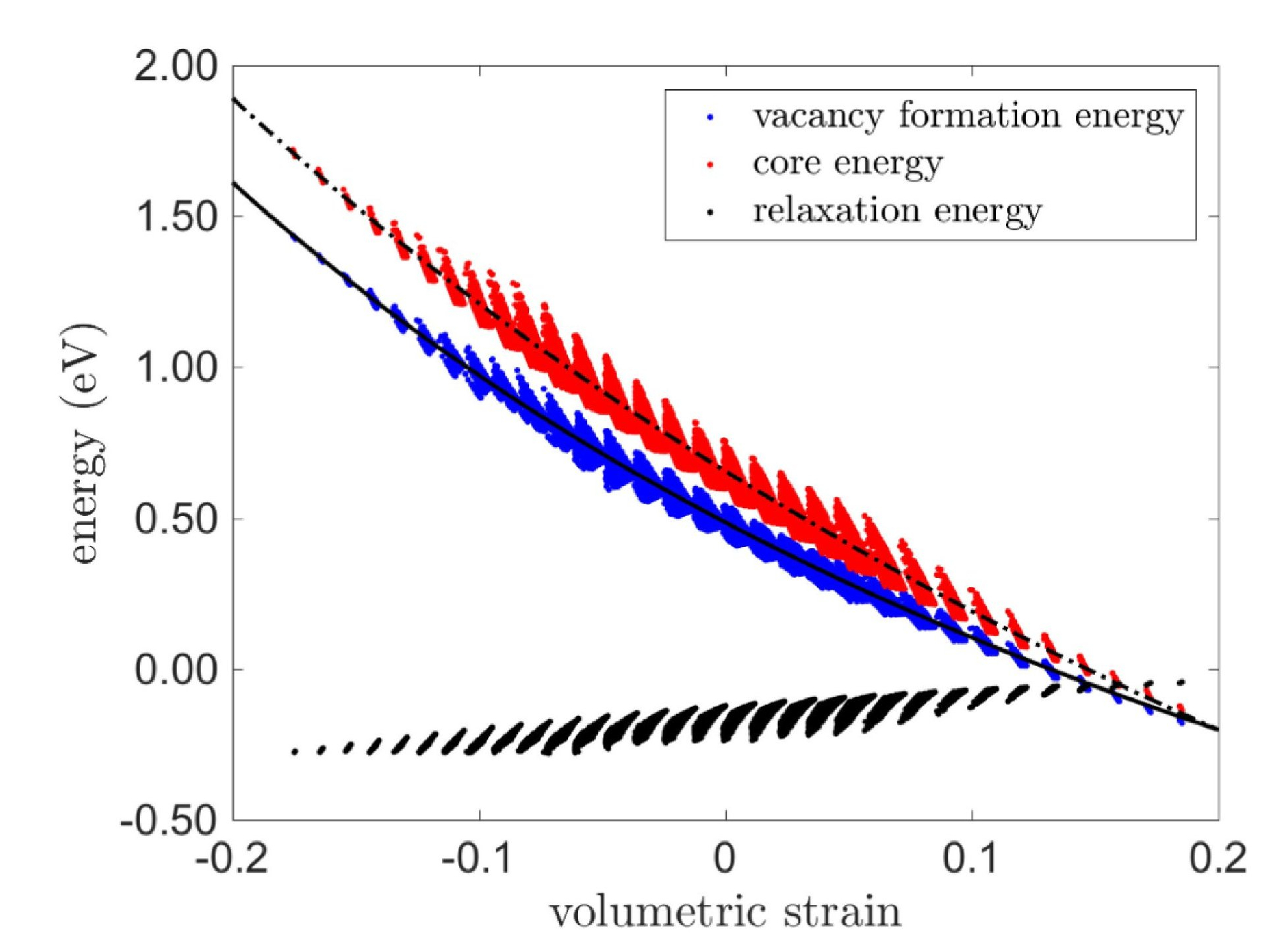
Aluminum: Vacancy formation energy
Electronic structure study regarding the influence of macroscopic deformations on the vacancy formation energy in aluminum
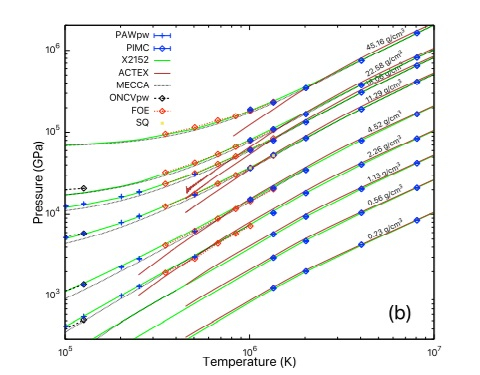
Equation of state: Boron nitride
Equation of state of warm-dense boron nitride combining computation, modeling, and experiment
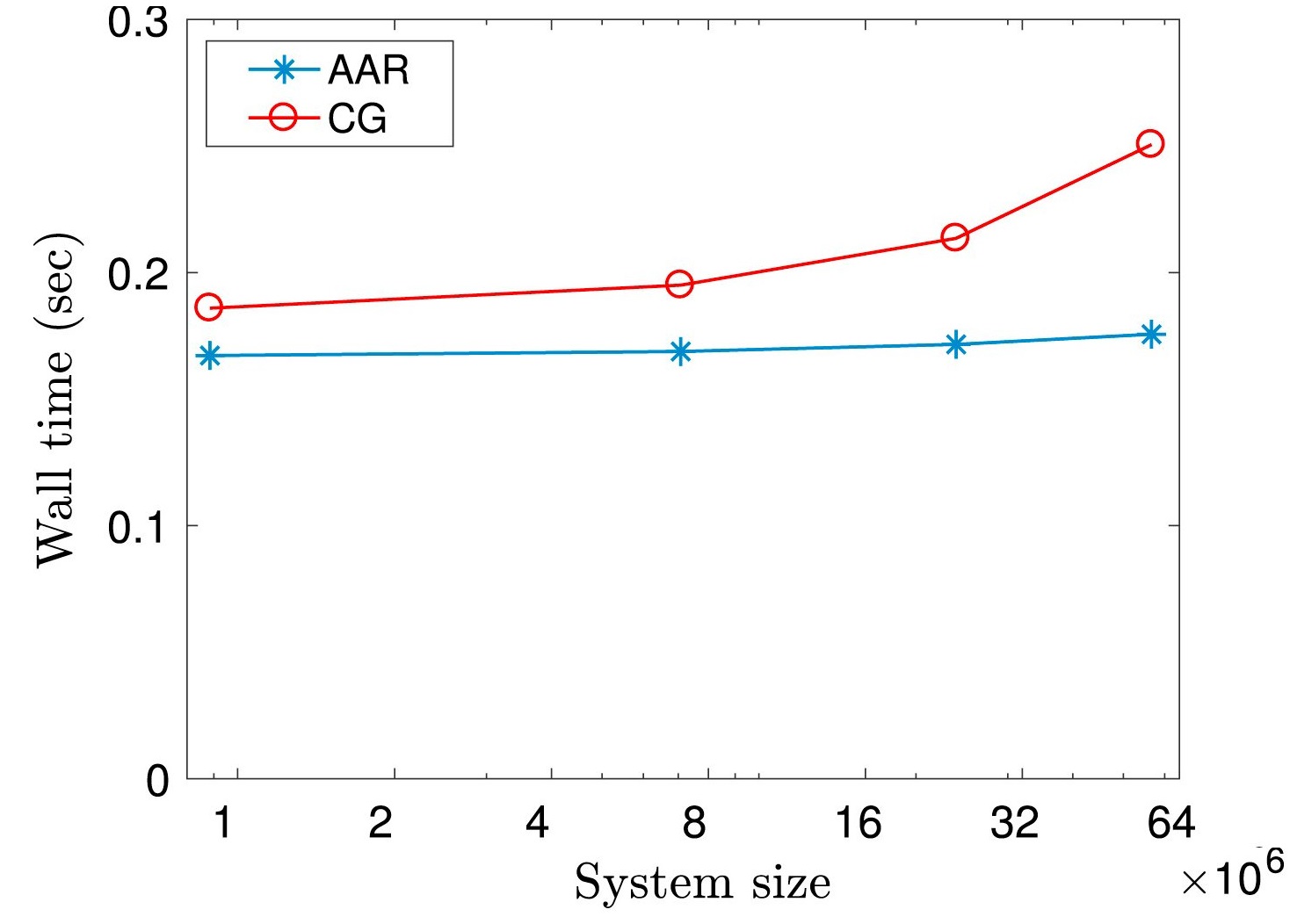
Linear solver: AAR II
Alternating Anderson–Richardson method: An efficient alternative to preconditioned Krylov methods for large, sparse linear systems
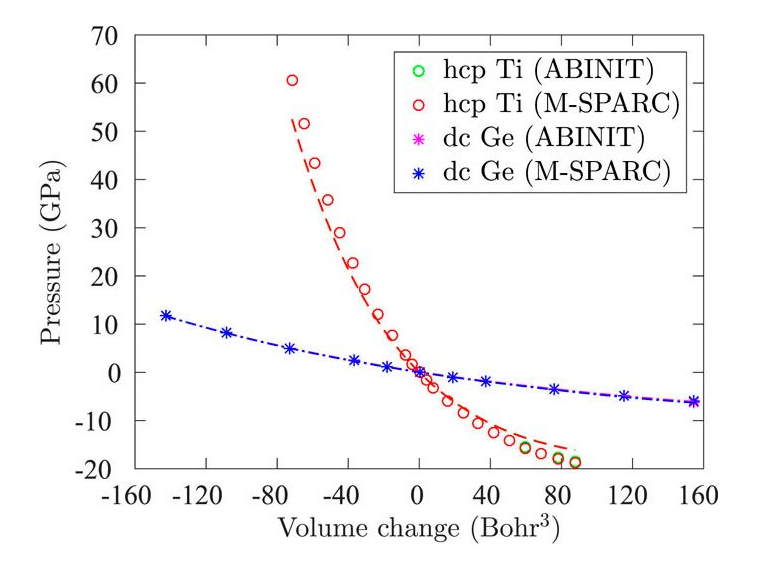
Kohn-Sham DFT: Stress tensor
On the calculation of the stress tensor in real-space Kohn-Sham density functional theory
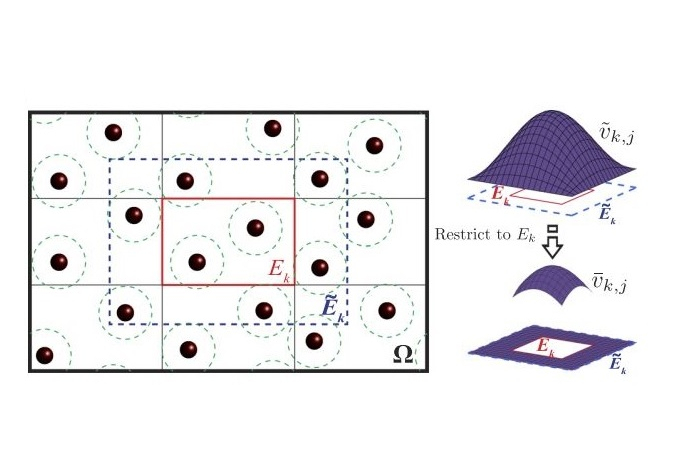
Kohn-Sham DFT: DDBP method
Discrete discontinuous basis projection method for large-scale electronic structure calculations
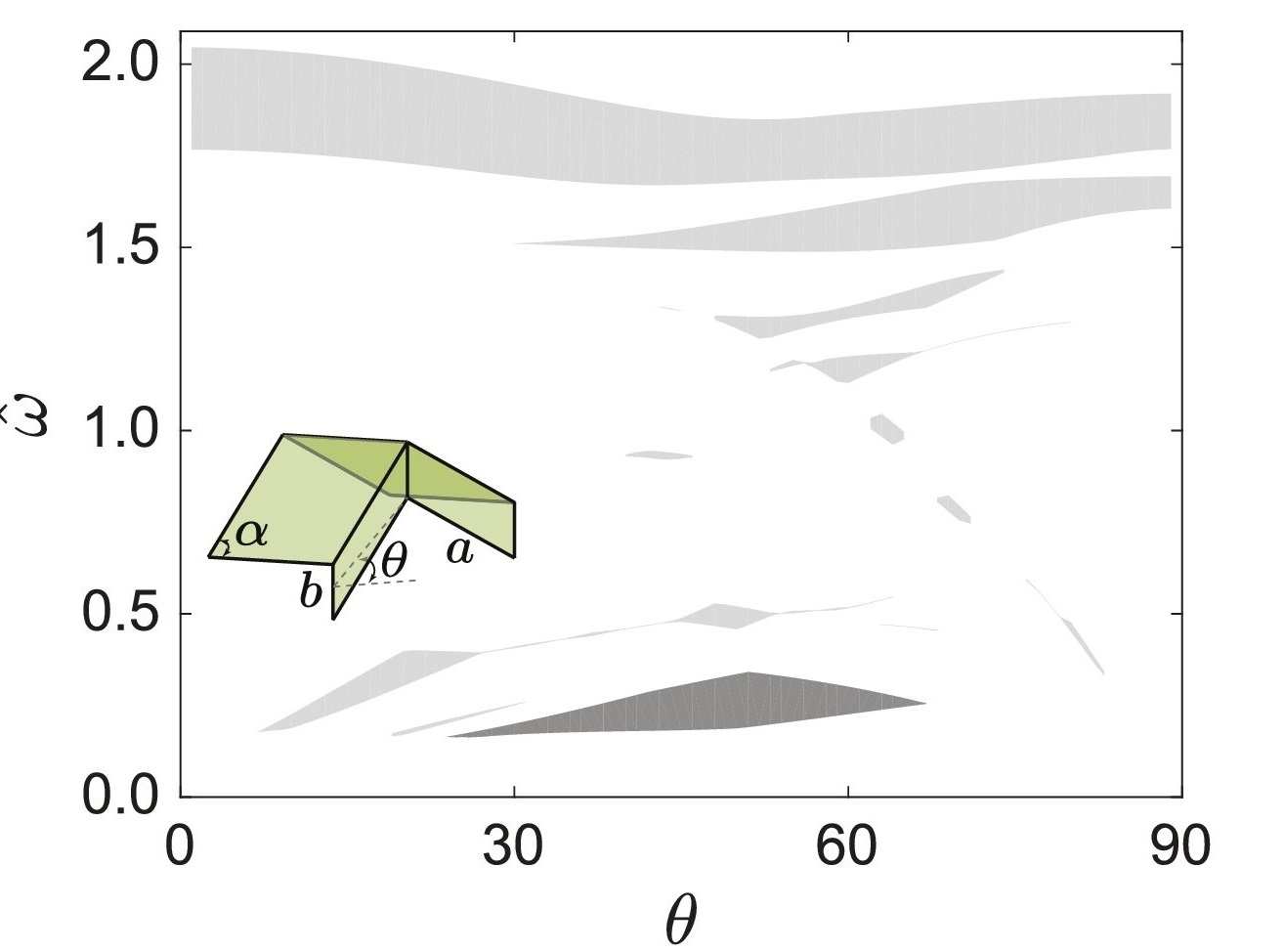
Origami: Bloch wave & Acoustic metamaterials
Bloch wave framework for structures with nonlocal interactions: Application to the design of origami acoustic metamaterials
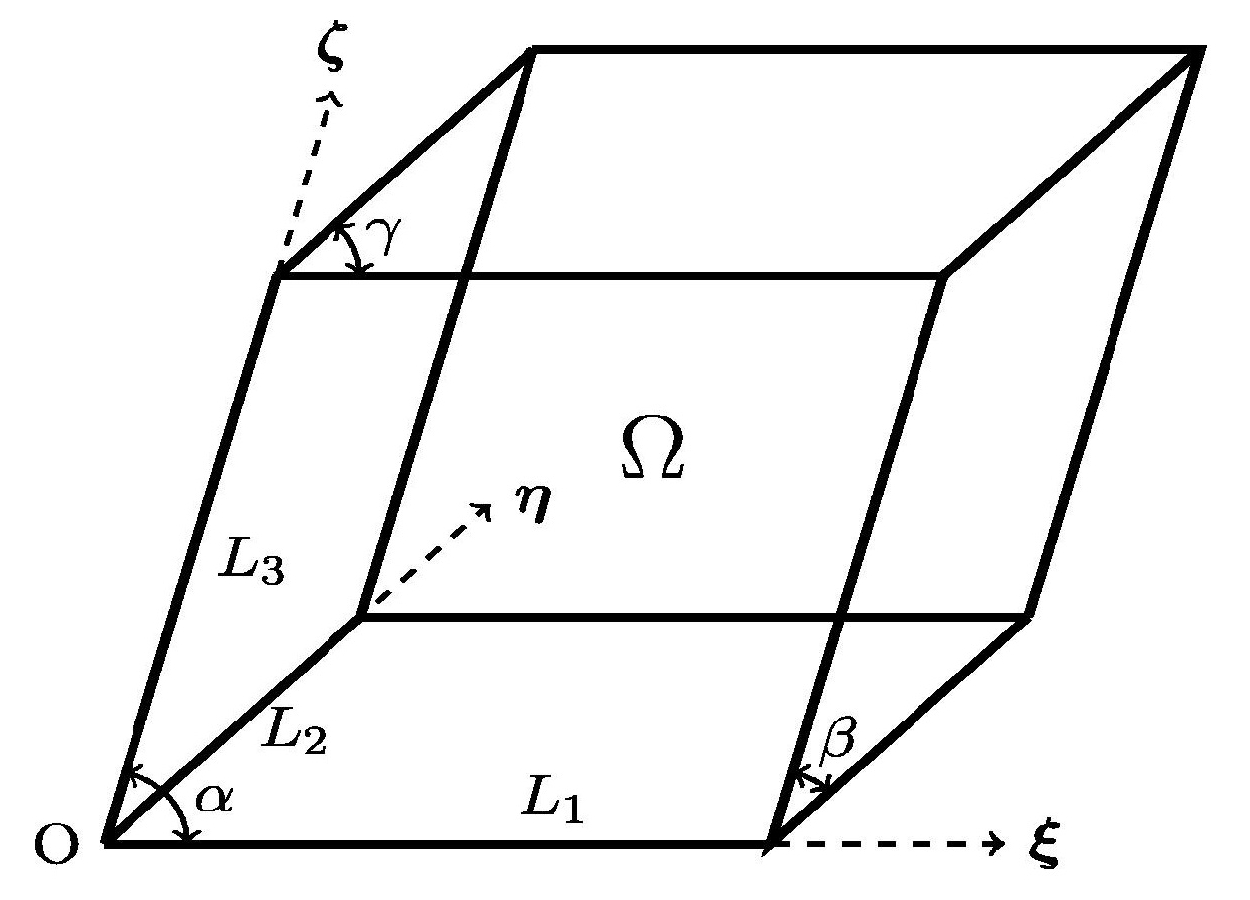
Kohn-Sham DFT: Non-orthogonal systems
On real-space Density Functional Theory for non-orthogonal crystal systems: Kronecker product formulation of the kinetic energy operator
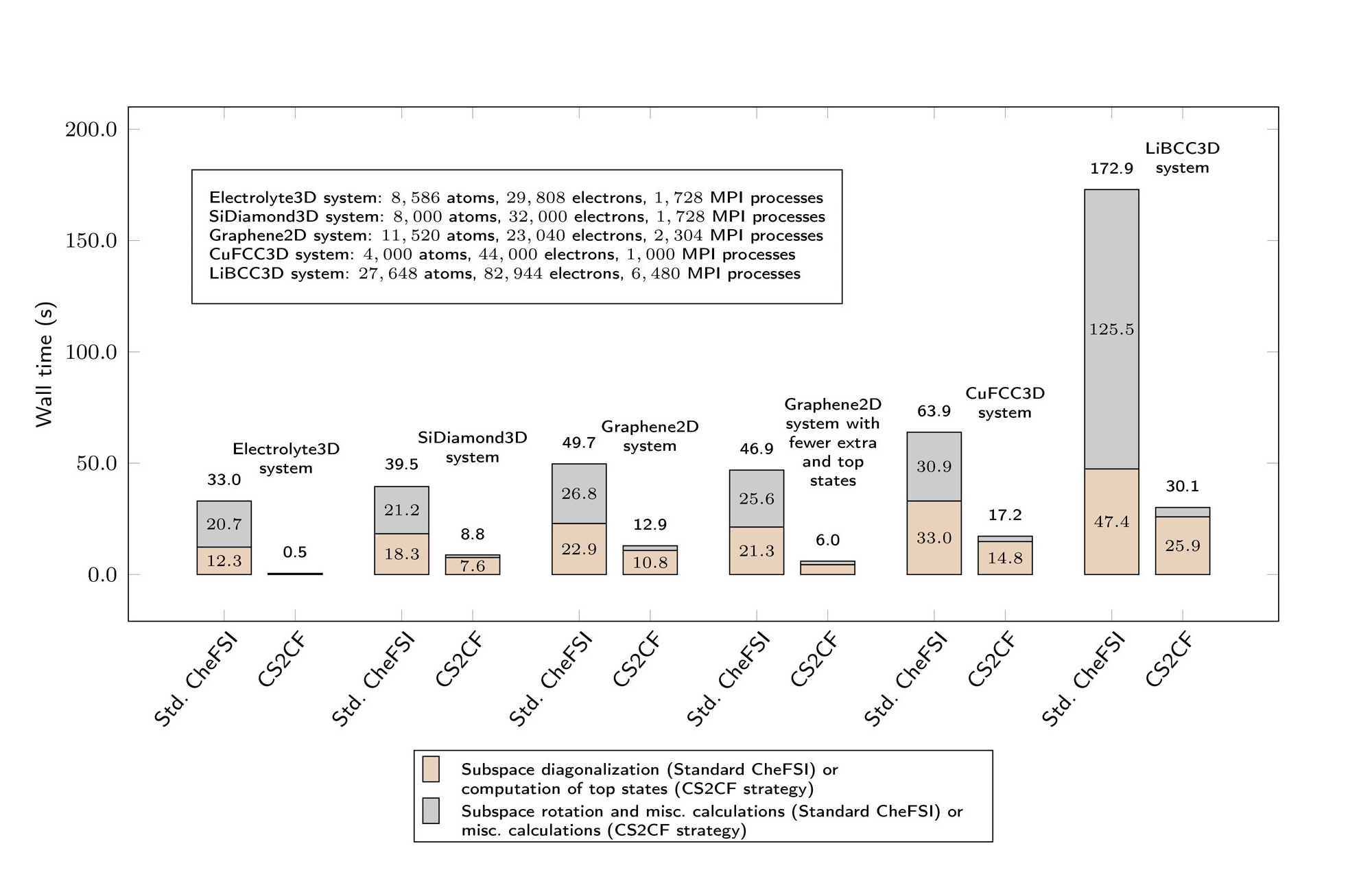
Kohn-Sham DFT: Complementary subspace method
Two-Level Chebyshev Filter Based Complementary Subspace Method: Pushing the Envelope of Large-Scale Electronic Structure Calculations
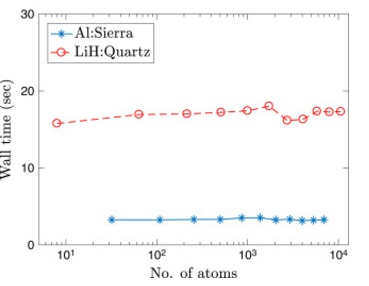
Kohn-Sham DFT: SQDFT
SQDFT: Spectral Quadrature method for large-scale parallel O(N) Kohn–Sham calculations at high temperature
Kohn-Sham DFT: SPARC II
SPARC: Accurate and efficient finite-difference formulation and parallel implementation of Density Functional Theory: Extended systems
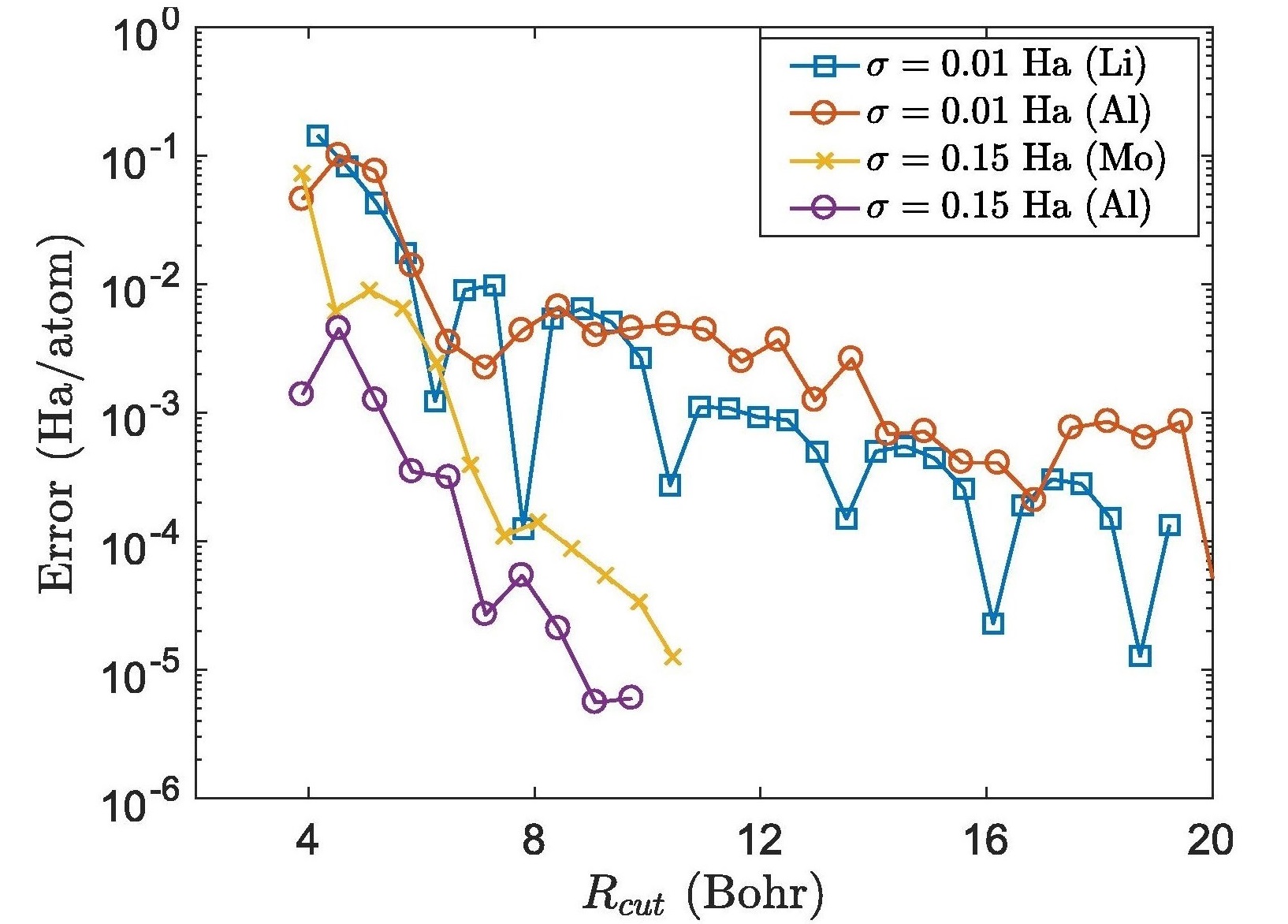
Kohn-Sham DFT: Nearsightedness in O(N) methods
On nearsightedness in metallic systems for Density Functional Theory calculations: A case study on aluminum
Kohn-Sham DFT: SPARC I
SPARC: Accurate and efficient finite-difference formulation and parallel implementation of Density Functional Theory: Isolated clusters
Instability prediction: Atomistic systems
On numerically predicting the onset and mode of instability in atomistic systems
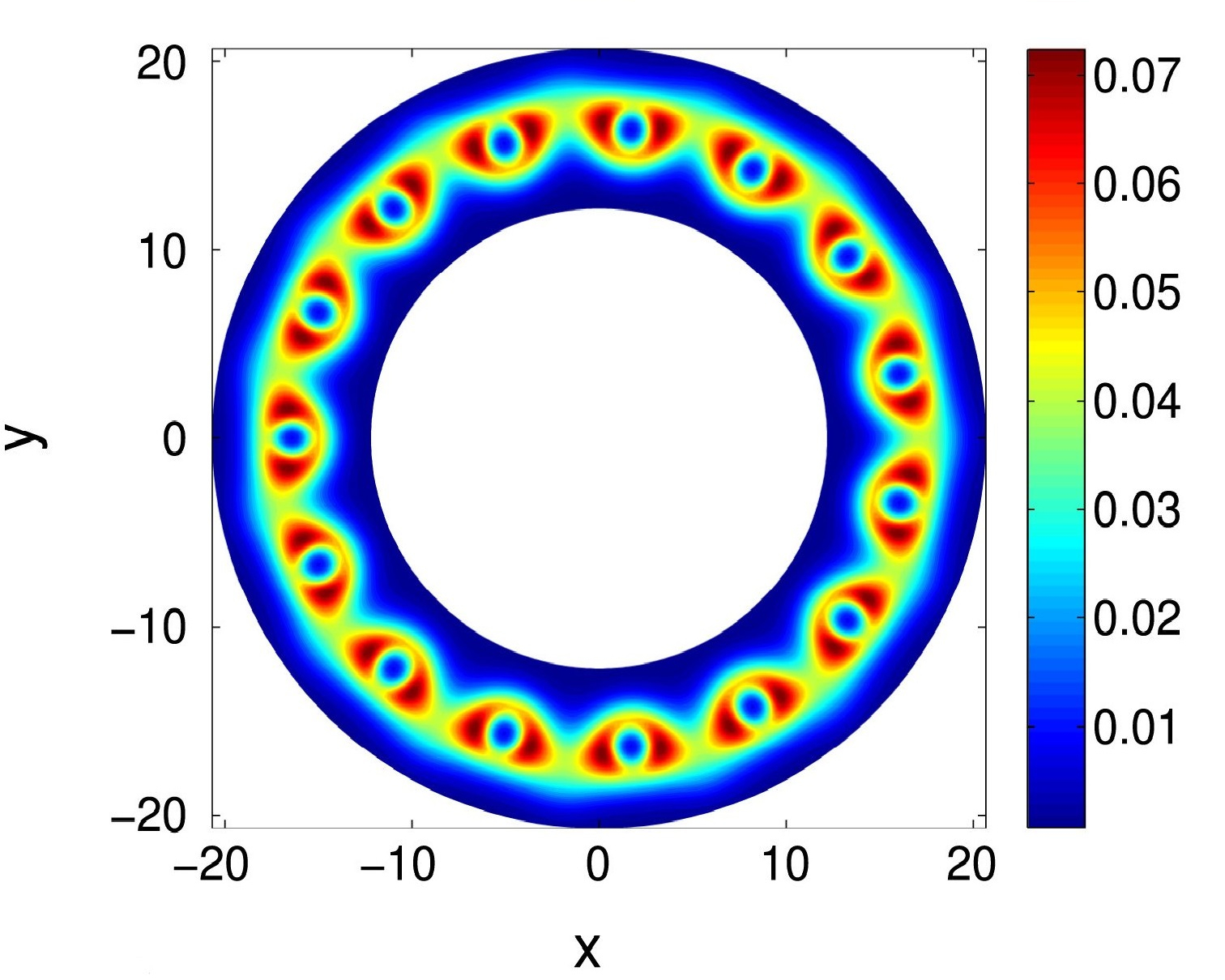
Kohn-Sham DFT: Cyclic DFT I
Cyclic density functional theory: A route to the first principles simulation of bending in nanostructures
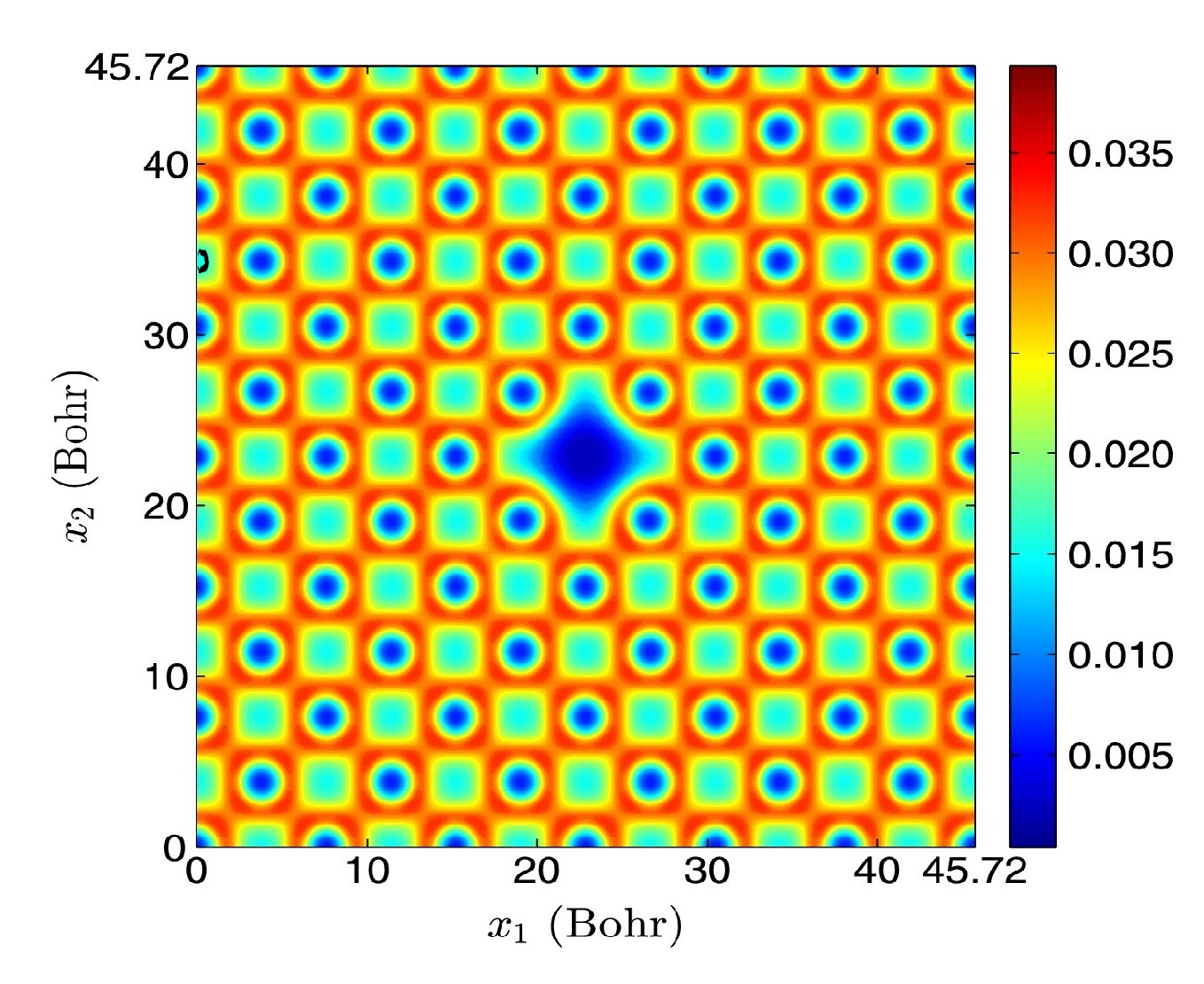
Orbital-free DFT: Periodic systems
Higher-order finite-difference formulation of periodic Orbital-free Density Functional Theory
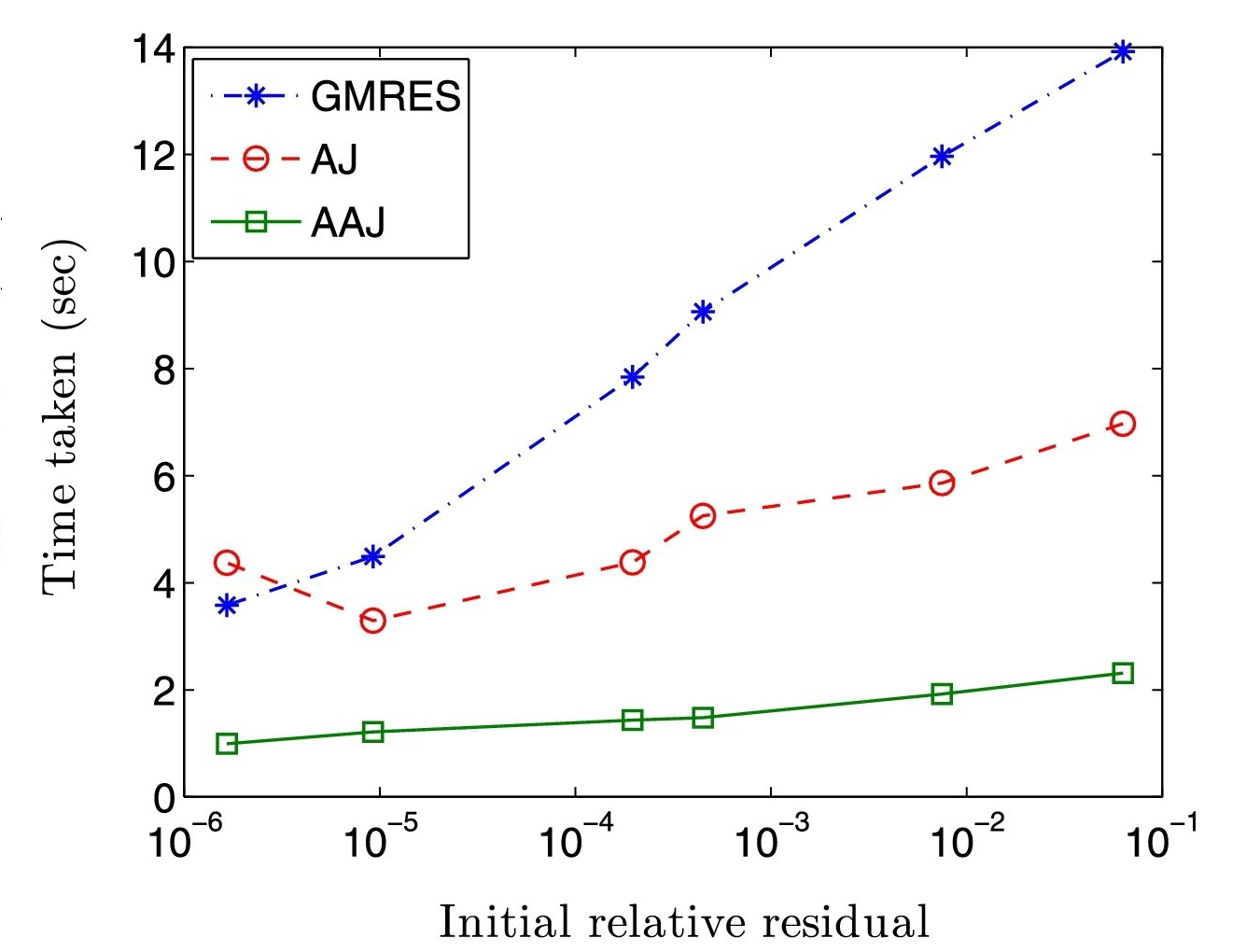
Linear solver: AAR I
Anderson acceleration of the Jacobi iterative method: An efficient alternative to Krylov methods for large, sparse linear systems
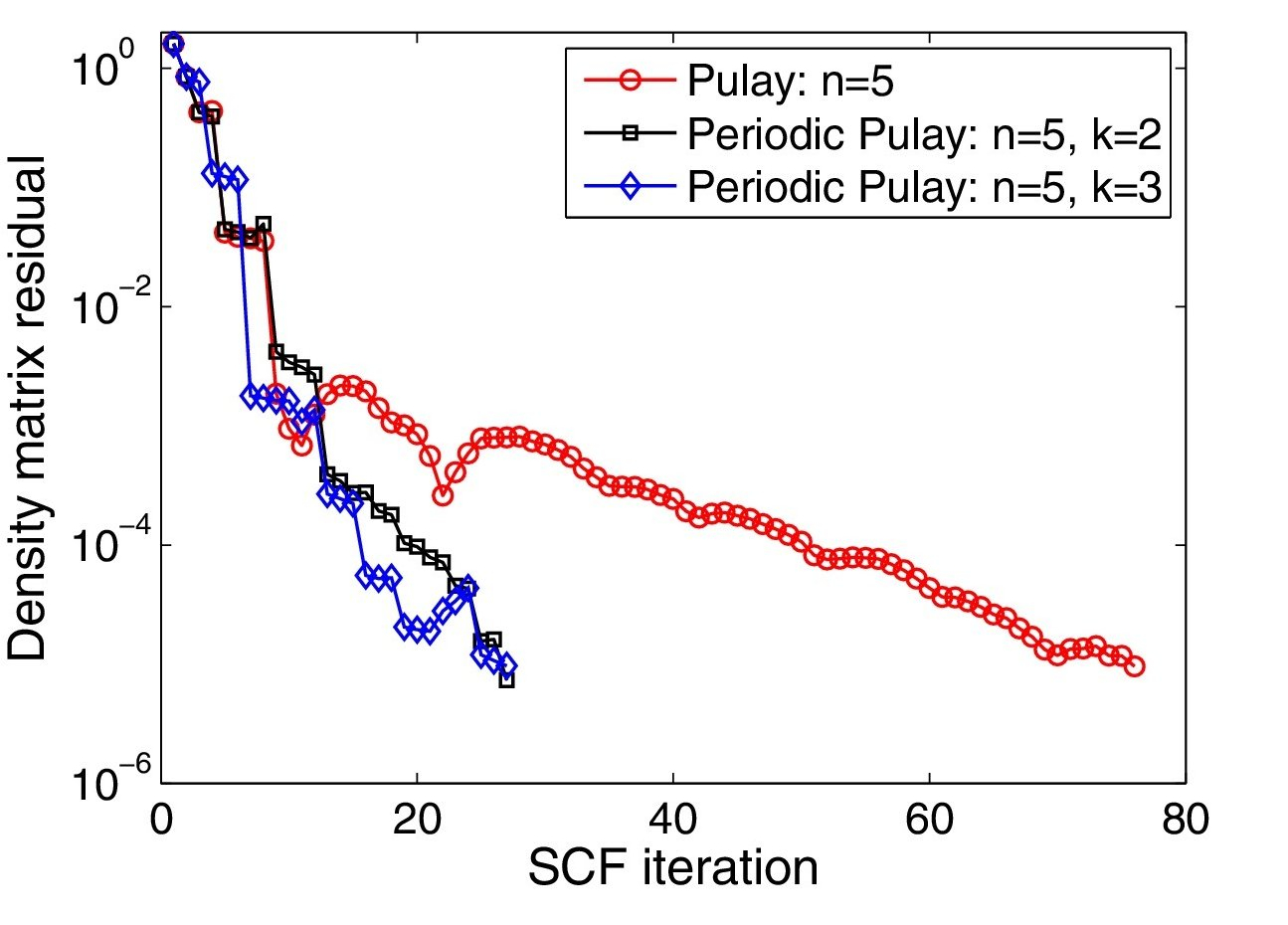
Kohn-Sham DFT: Periodic Pulay mixing
Periodic Pulay method for robust and efficient convergence acceleration of self-consistent field iterations
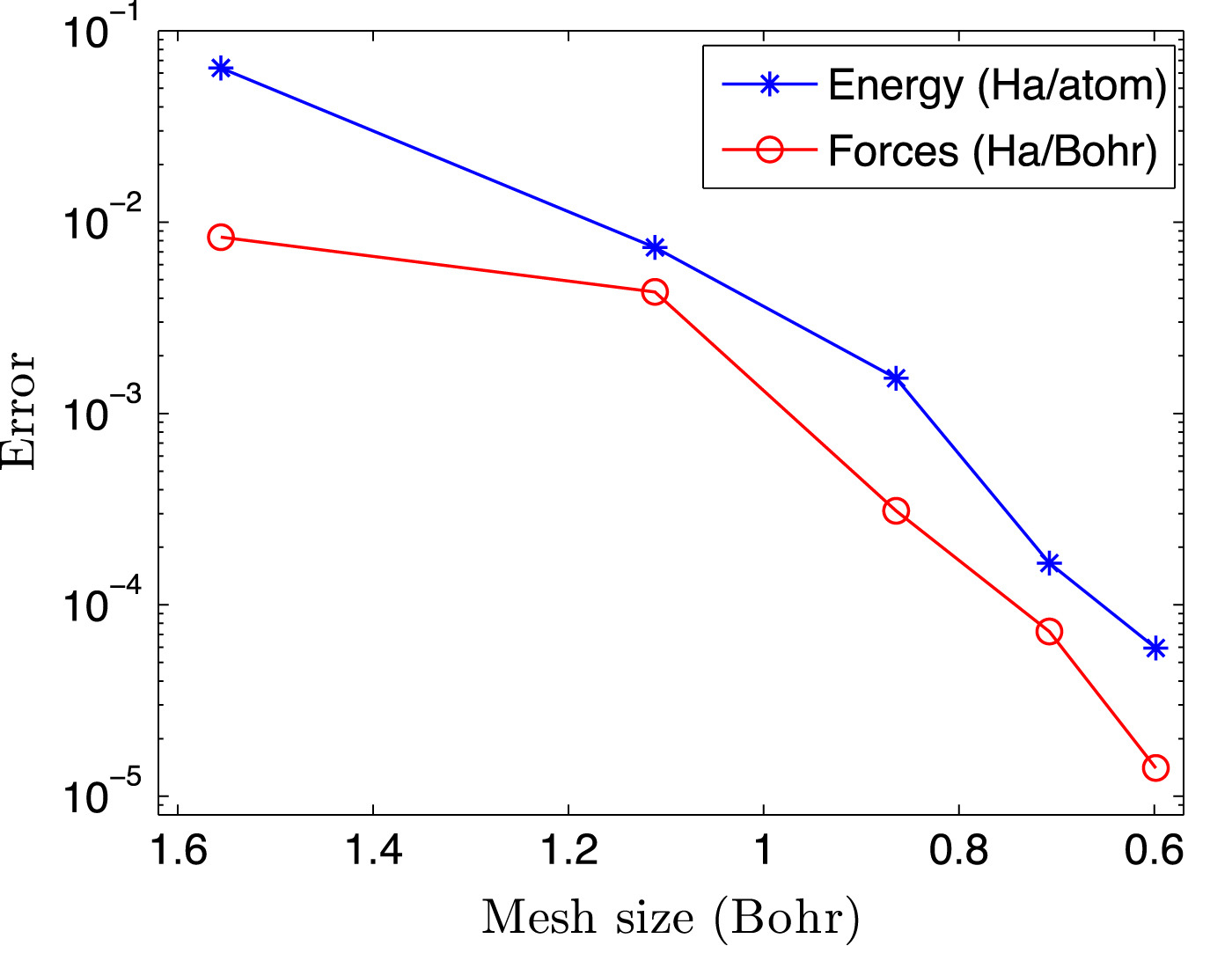
Kohn-Sham DFT: O(N) Spectral Quadrature
Spectral Quadrature method for accurate O(N) electronic structure calculations of metals and insulators
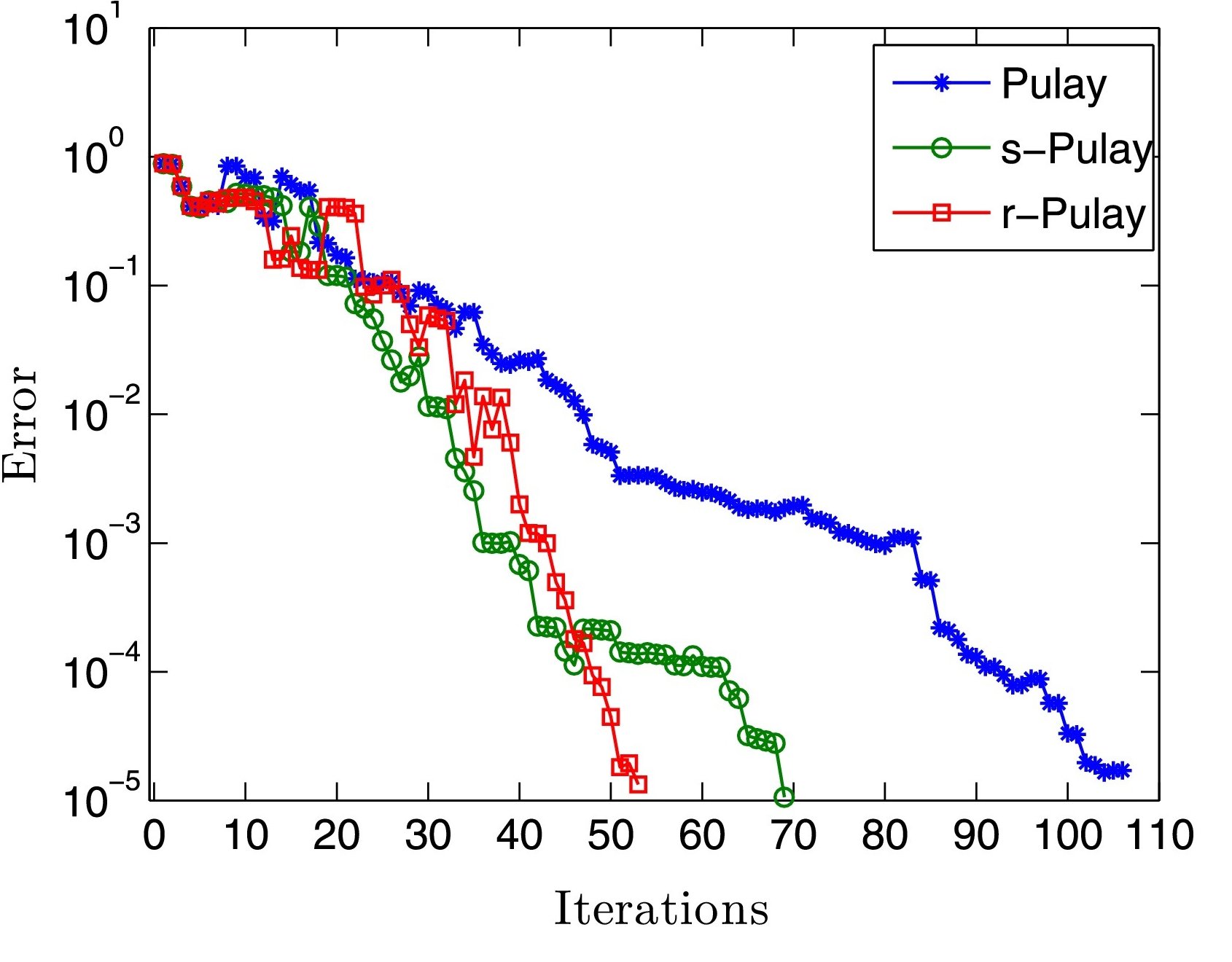
Kohn-Sham DFT: r-Pulay mixing
Restarted Pulay mixing for efficient and robust acceleration of fixed-point iterations
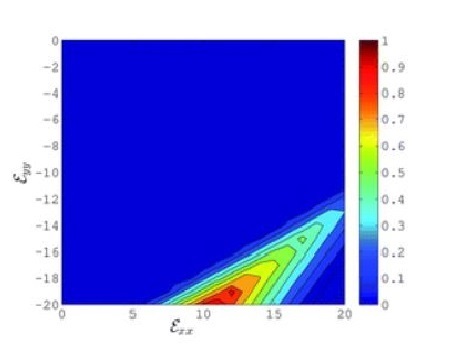
Graphene: Strain engineering
Ab initio strain engineering of graphene: opening bandgaps up to 1 eV
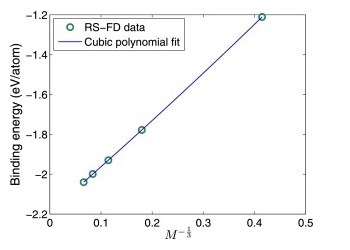
Orbital-free DFT: Non periodic systems
Augmented Lagrangian formulation of orbital-free density functional theory
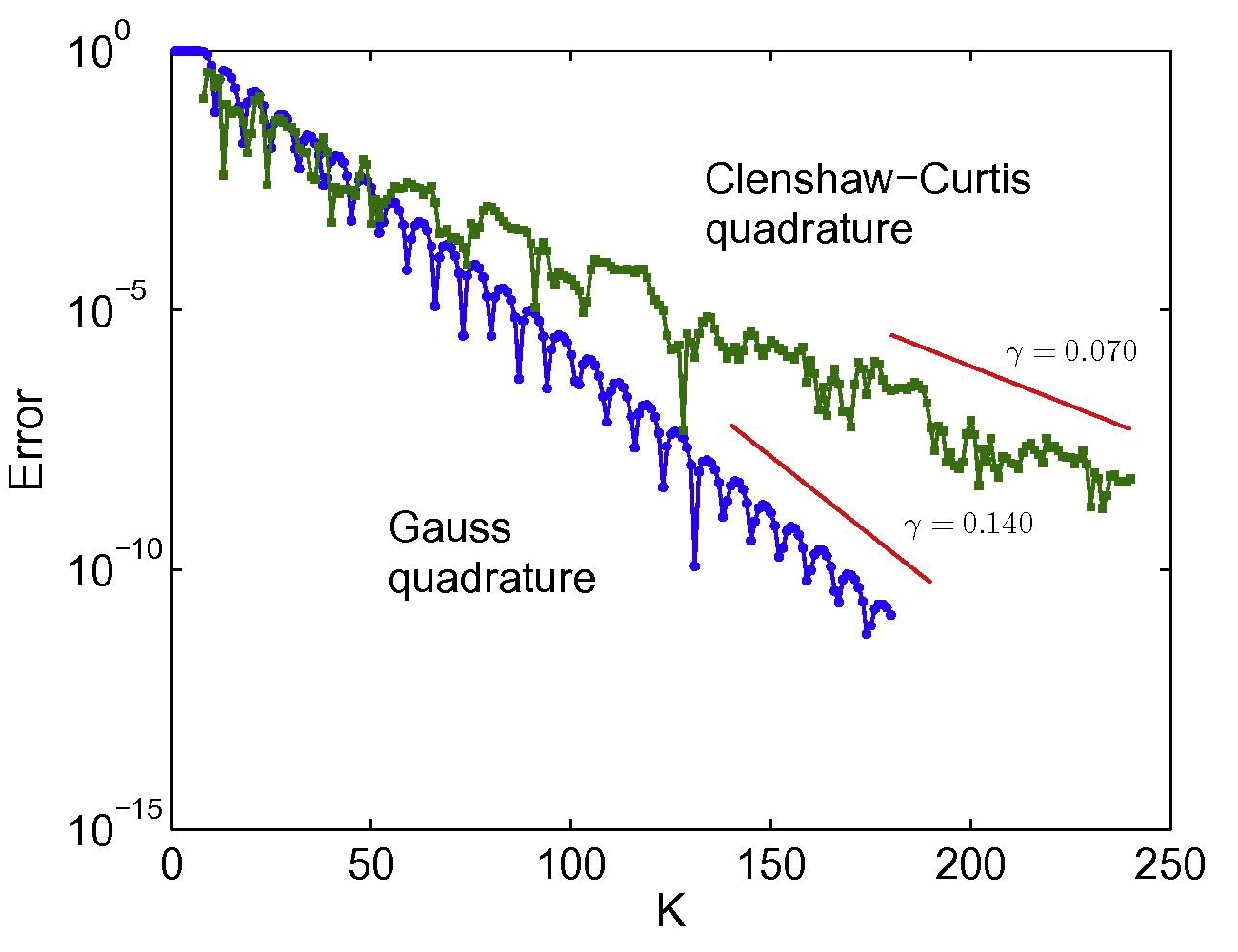
Kohn-Sham DFT: O(N) Spectral Quadrature
On spectral quadrature for linear-scaling Density Functional Theory
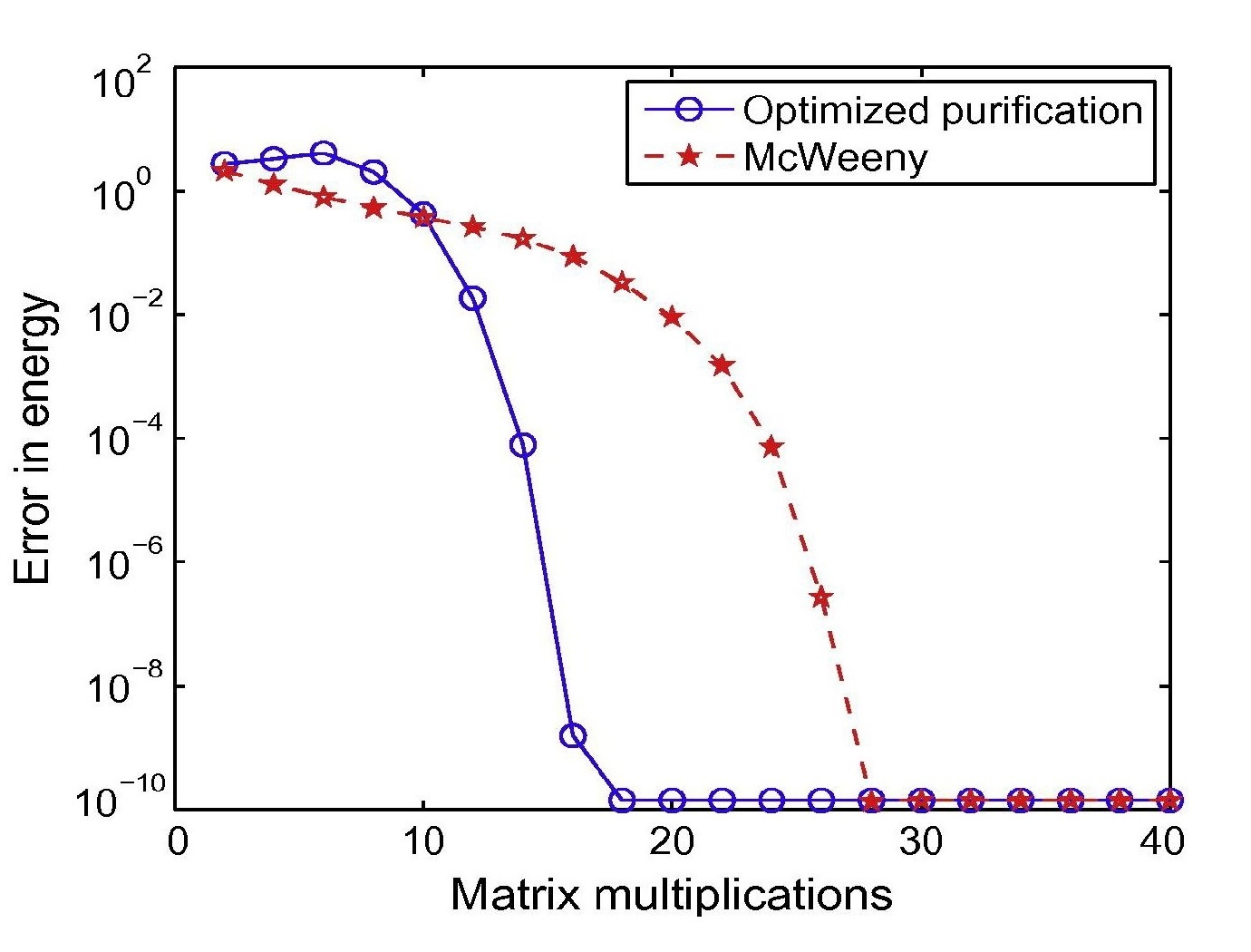
Kohn-Sham DFT: O(N) optimized Purification
Optimized purification for density matrix calculation
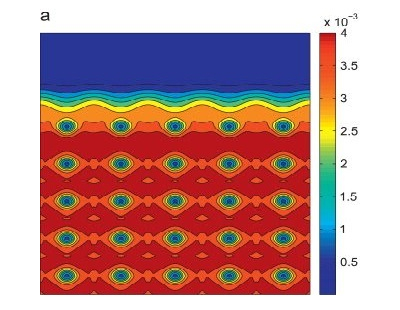
Kohn-Sham DFT: Coarse-graining
Coarse-graining Kohn–Sham Density Functional Theory
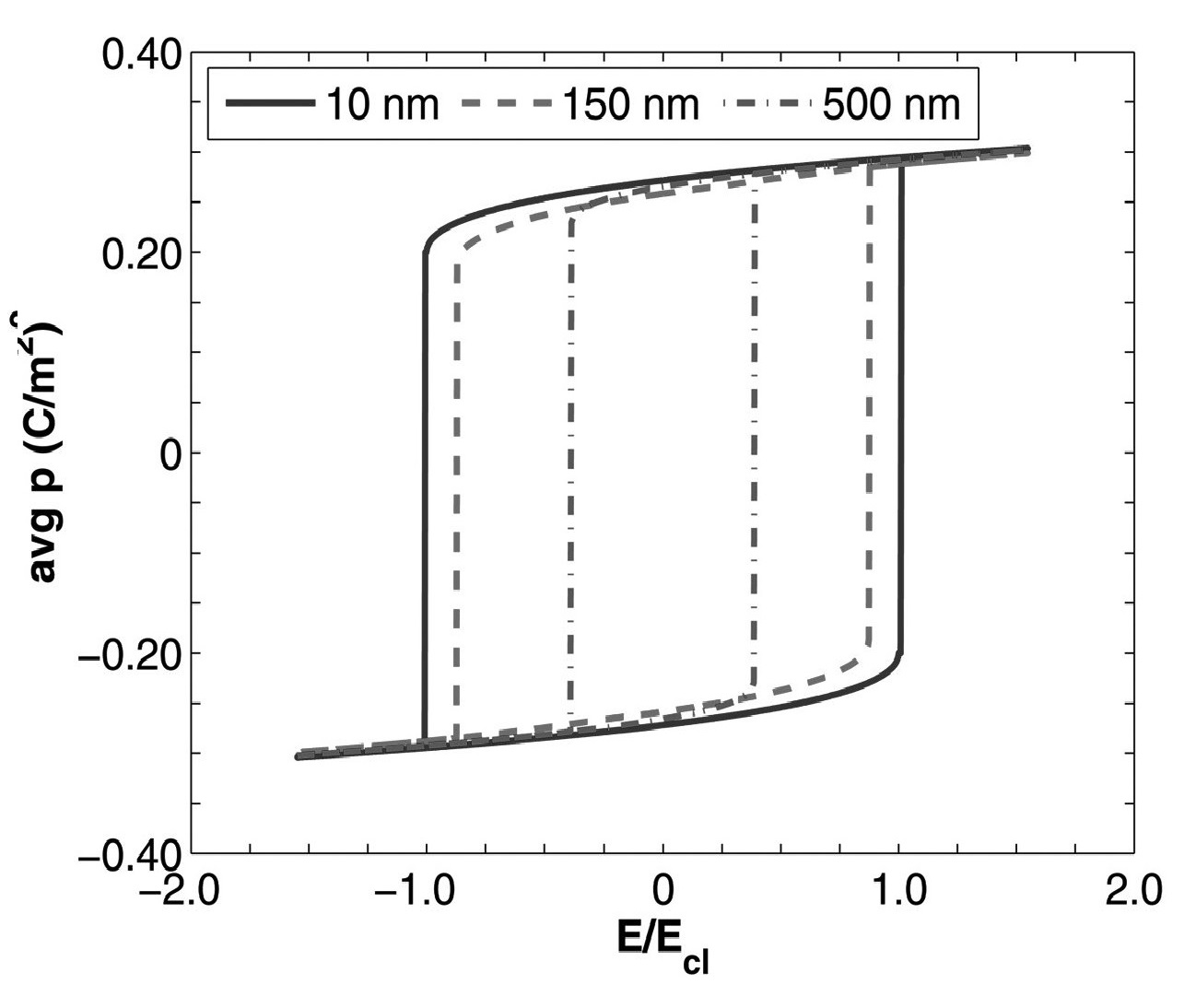
Ferroelectrics: Semiconductor model
Evolution of polarization and space charges in semiconducting ferroelectrics
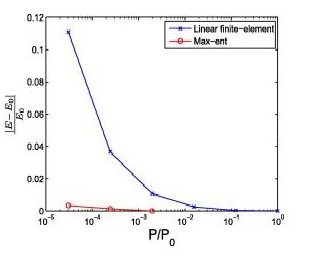
Kohn-Sham DFT: Mesh free discretization
A mesh-free convex approximation scheme for Kohn–Sham density functional theory

Kohn-Sham DFT: Finite element discretization
Non-periodic finite-element formulation of Kohn–Sham density functional theory
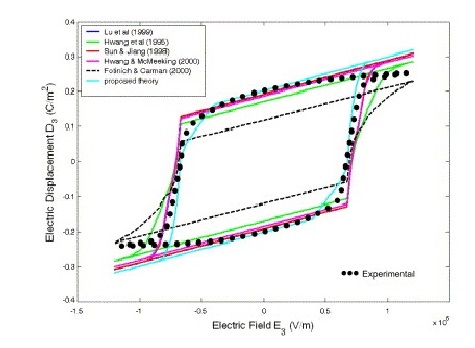
Ferroelectrics: Domain switching criteria I
Domain switching criteria for ferroelectrics
CONFERENCE/WORKSHOP
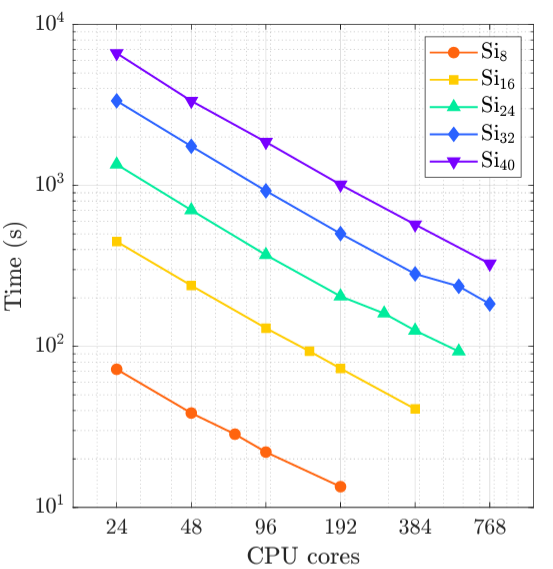
Kohn-Sham DFT: RPA correlation energy
Many-Body Electronic Correlation Energy using Krylov Subspace Linear Solvers
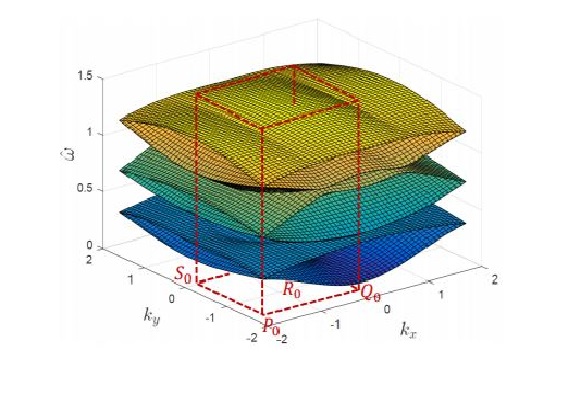
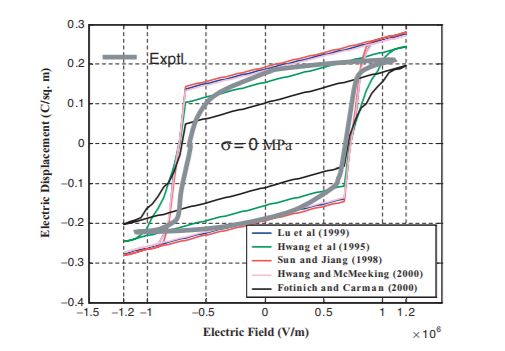
Ferroelectrics: Domain switching criteria II
Domain Switching Criteria for Tetragonal Phase Ferroelectrics: A Comparative Study
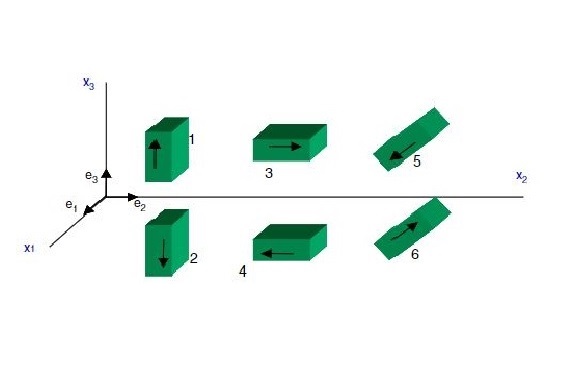
Miscellaneous
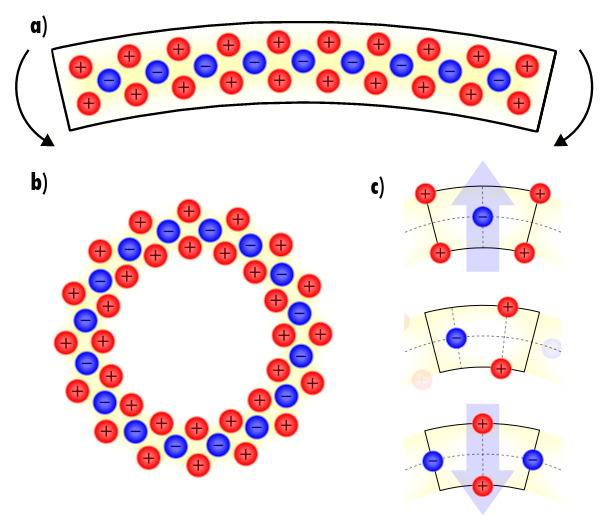
Perspective: 2D flexoelectricity
Towards understanding flexoelectricity at the nanoscale
PREPRINT
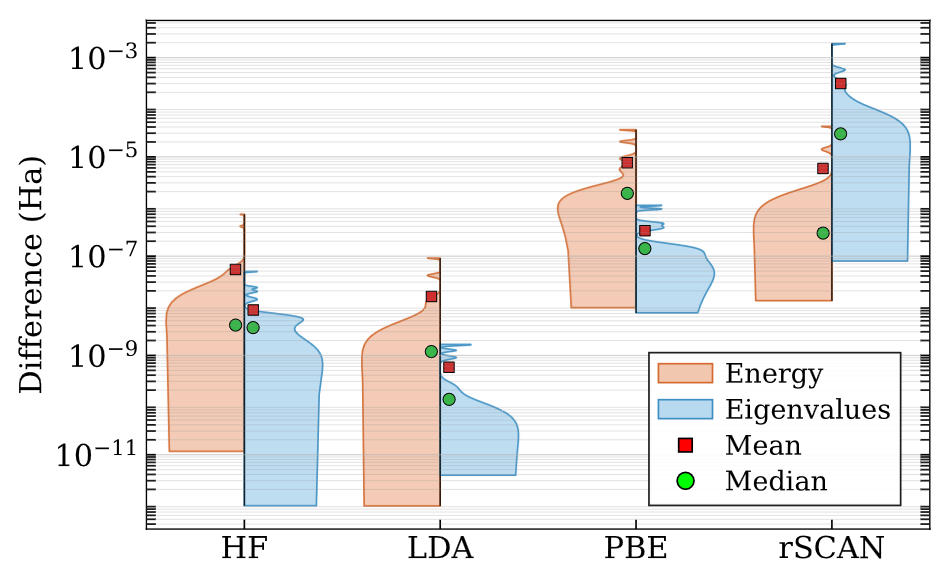
Kohn-Sham DFT: SPARC-atomSFE v1.0
SPARC-atomSFE: Spectral finite-element package for atomic structure calculations in density functional theory
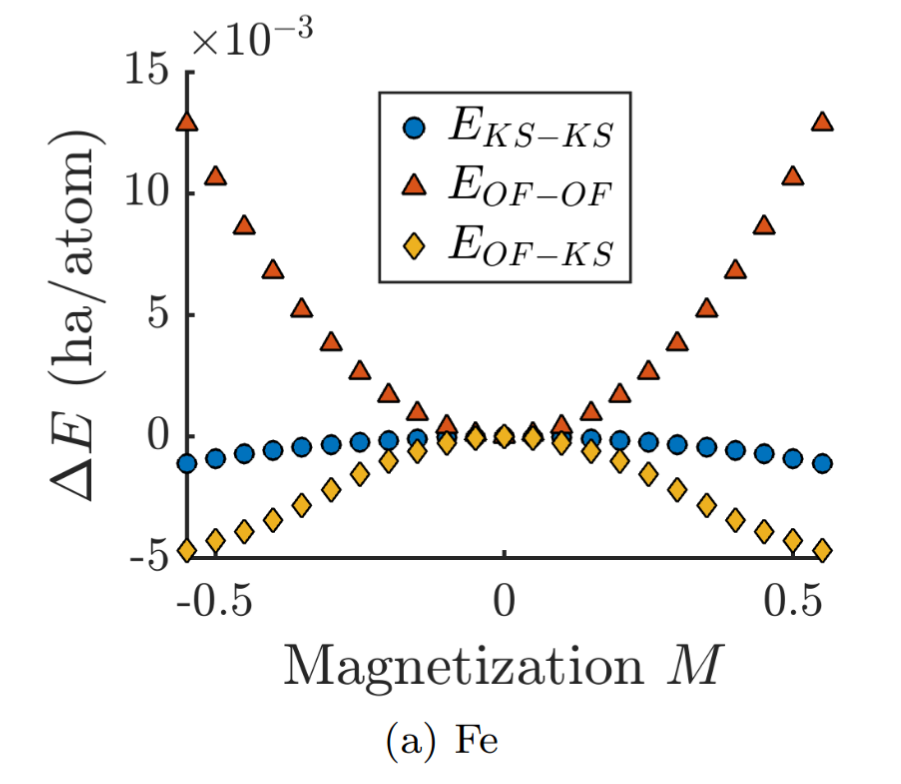
Orbital-free DFT: Itinerant magnetism
Assessing the suitability of the Thomas-Fermi-von Weizsäcker density functional for itinerant magnetism
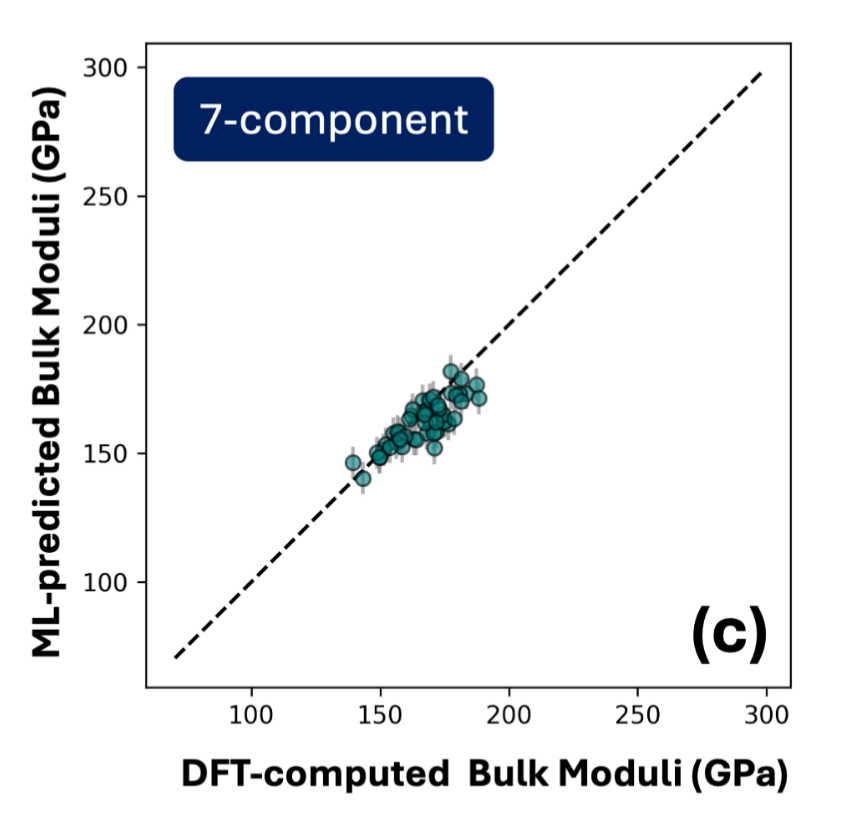
ML-DFT: High Entropy Alloys
Universal electronic manifolds for extrapolative alloy discovery
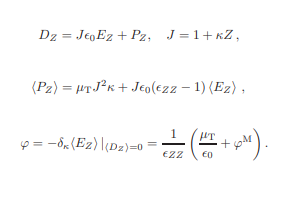
Note on transversal flexoelectricity
A note on transversal flexoelectricity in two-dimensional systems
arXiv (2022)