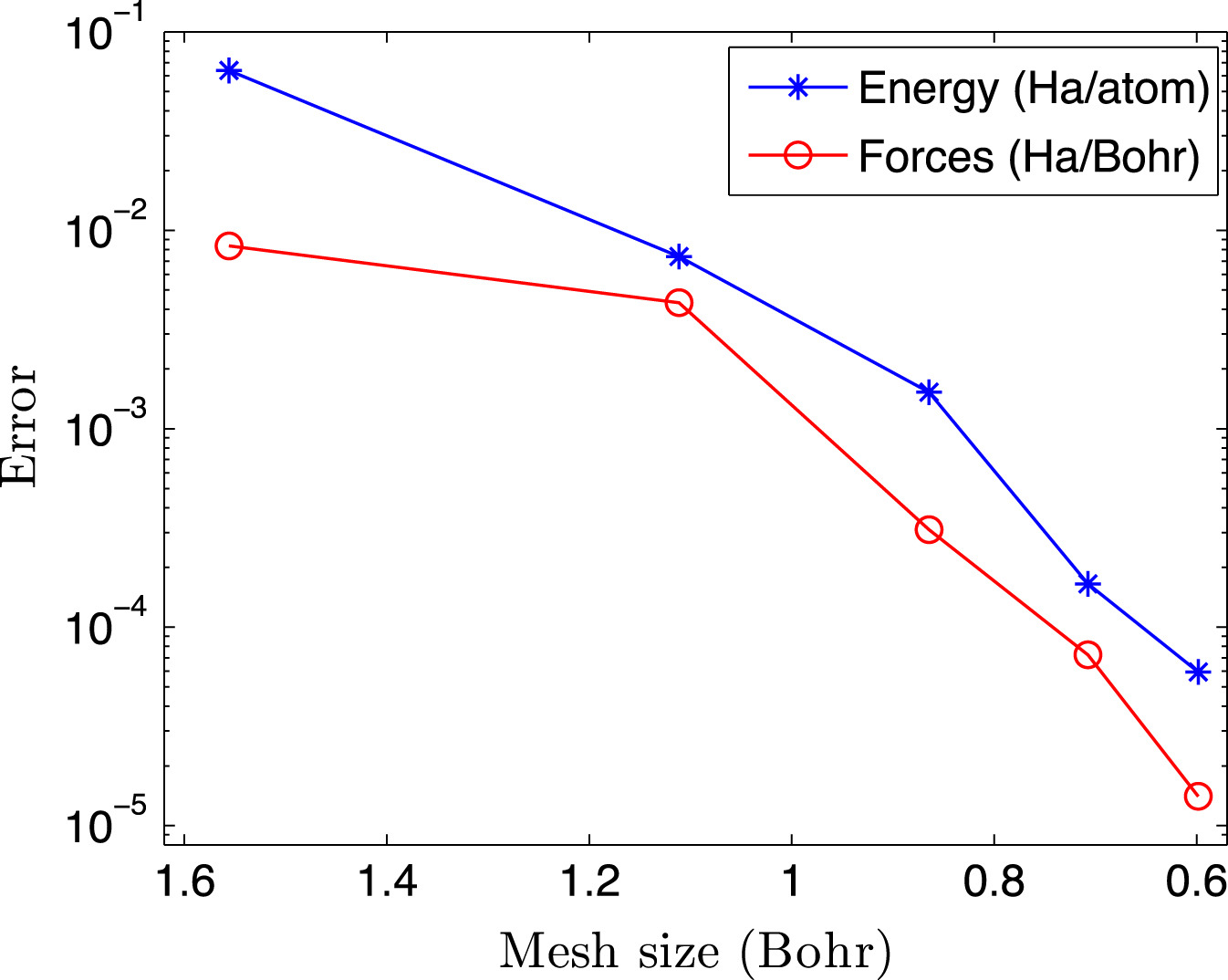
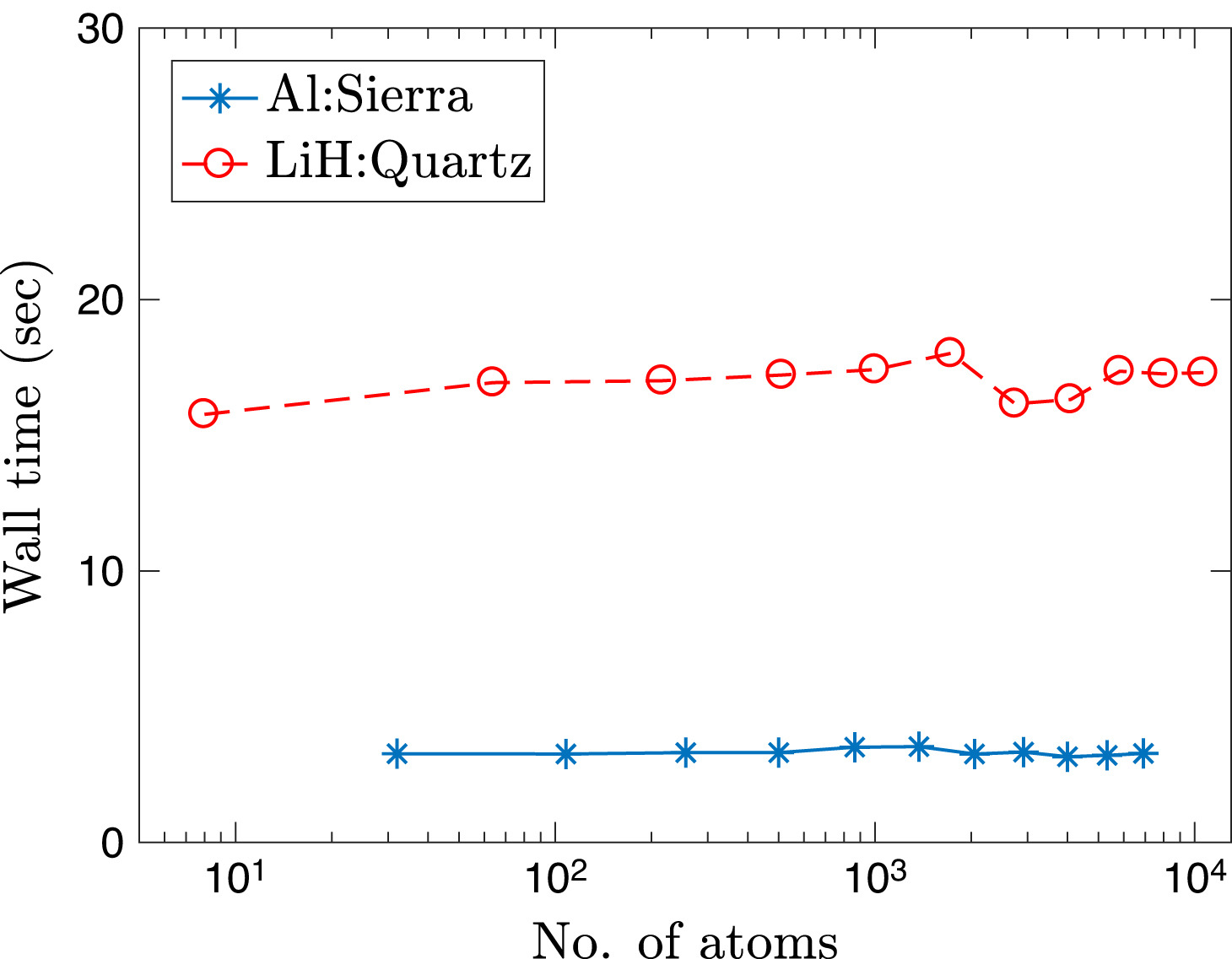
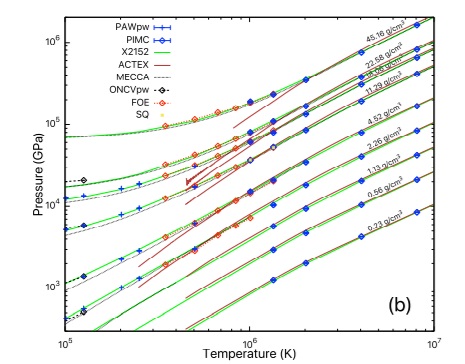
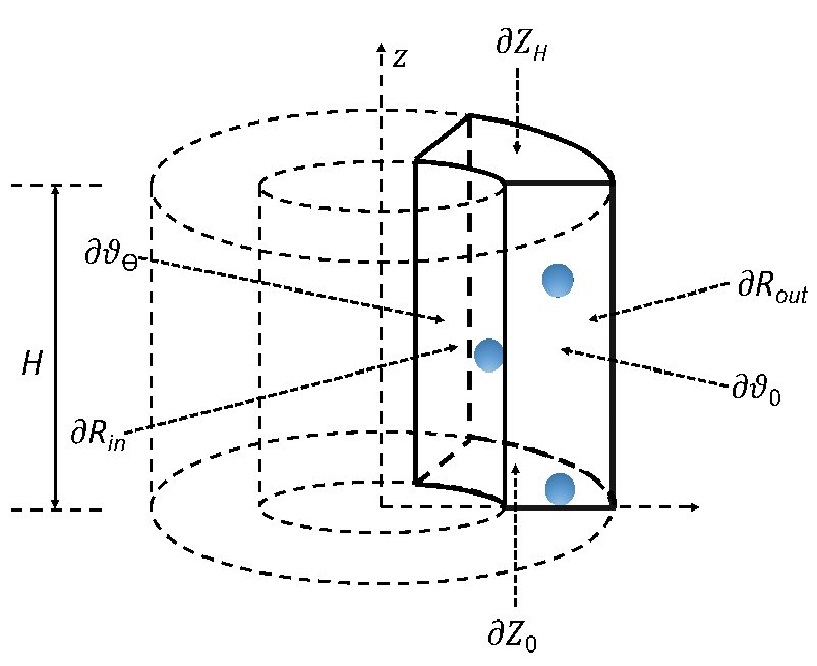
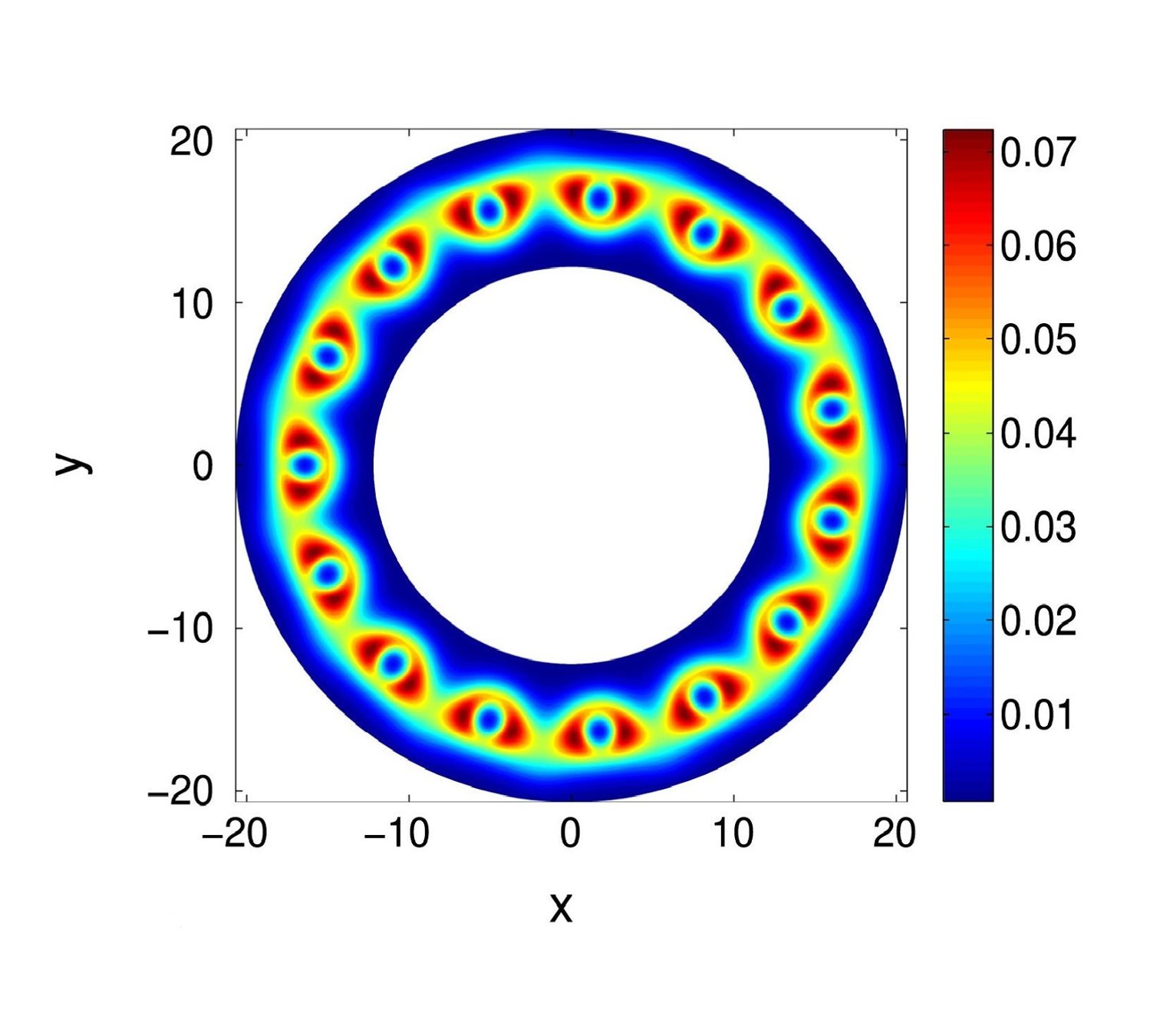
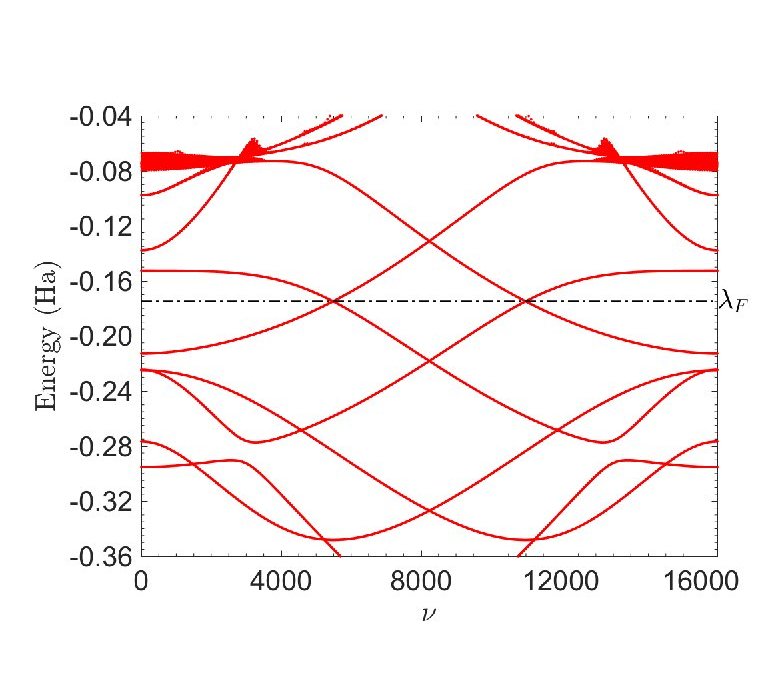
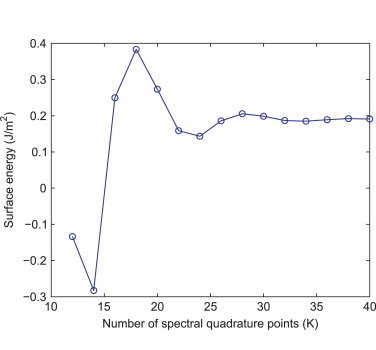
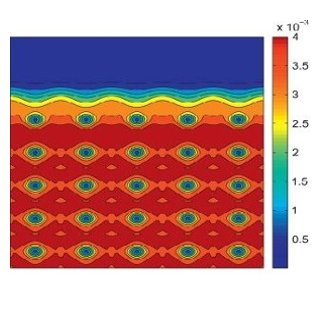
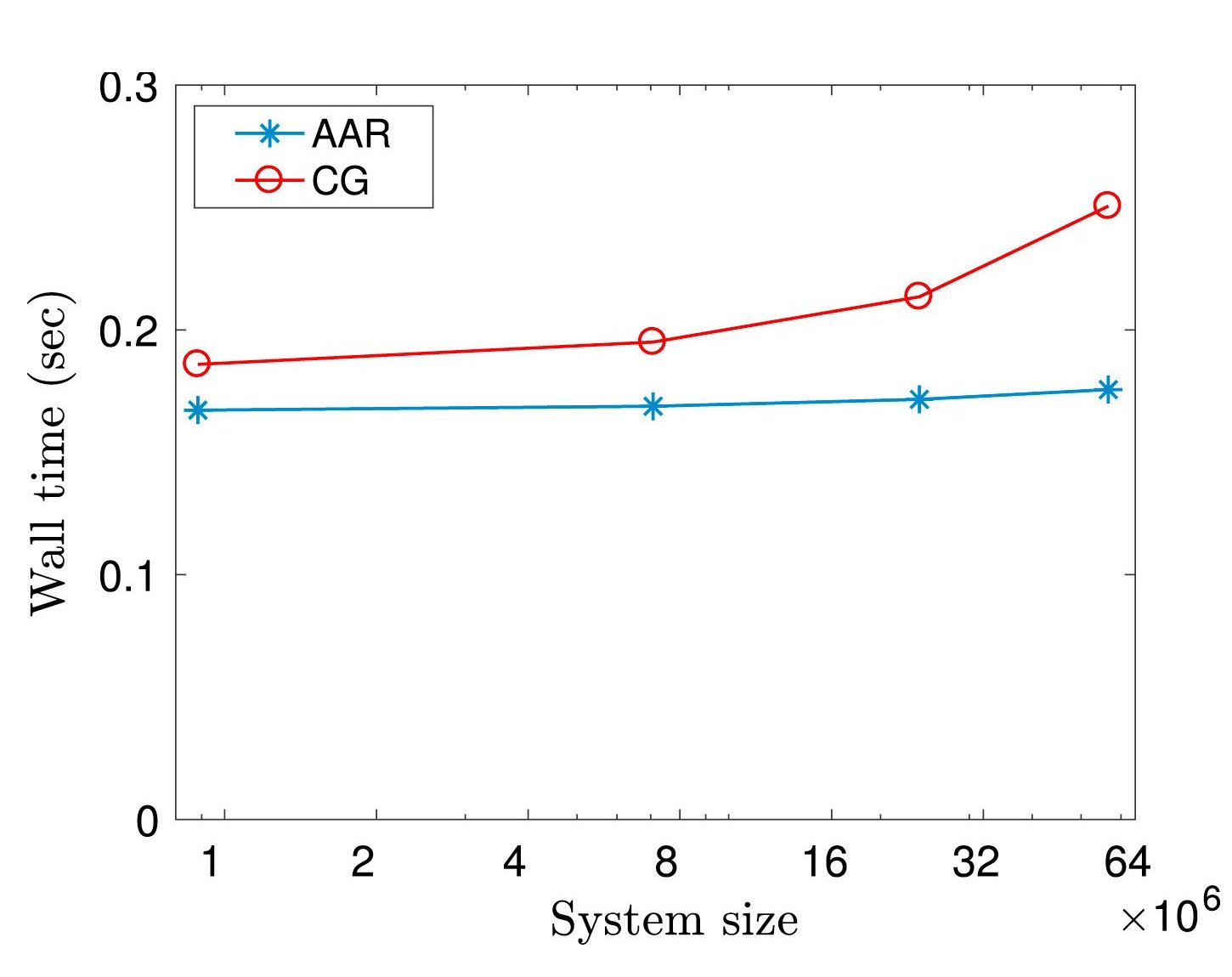
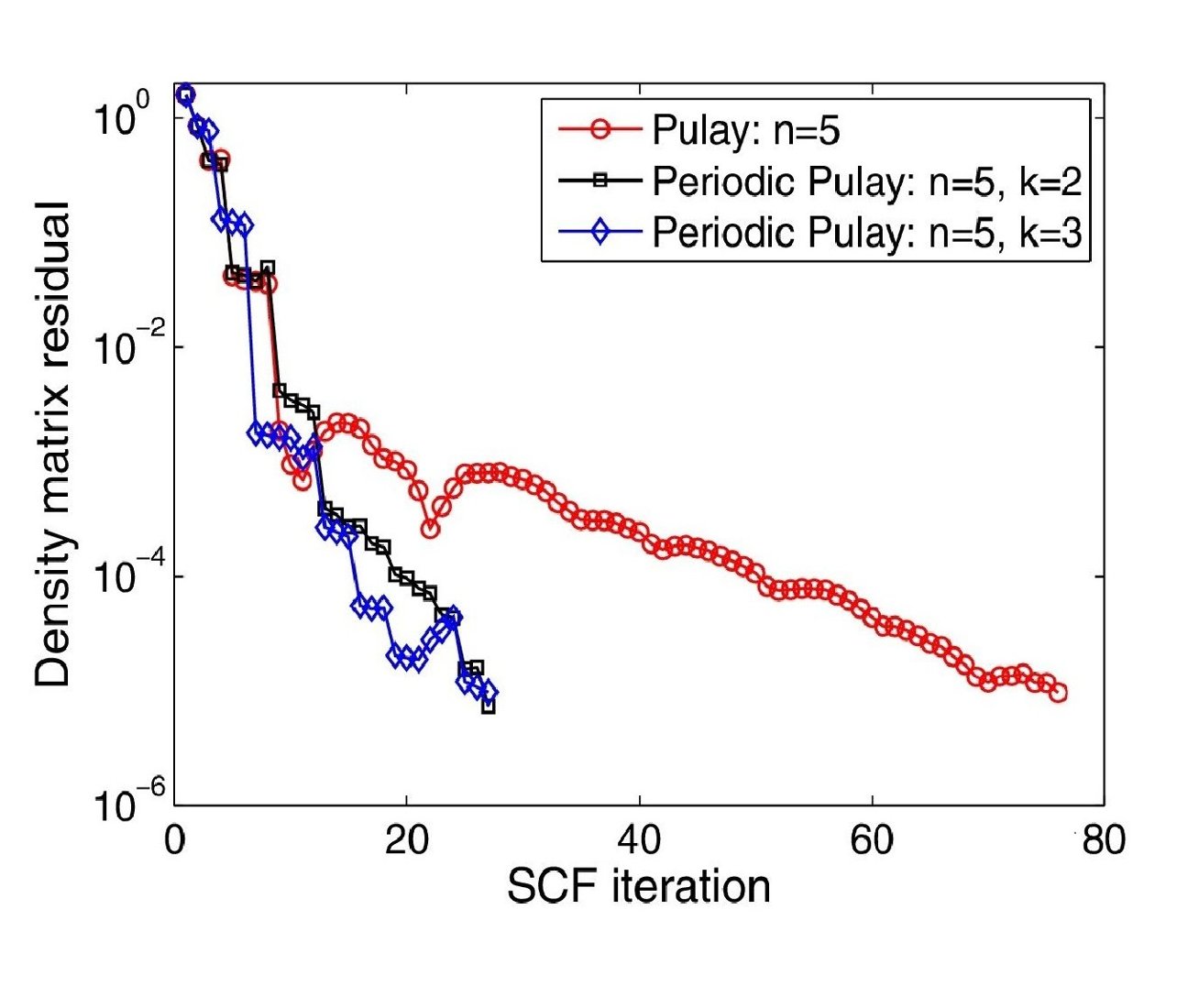
Ⅰ. Simulation Package for Ab-initio Real-space Calculations at Extreme Scales
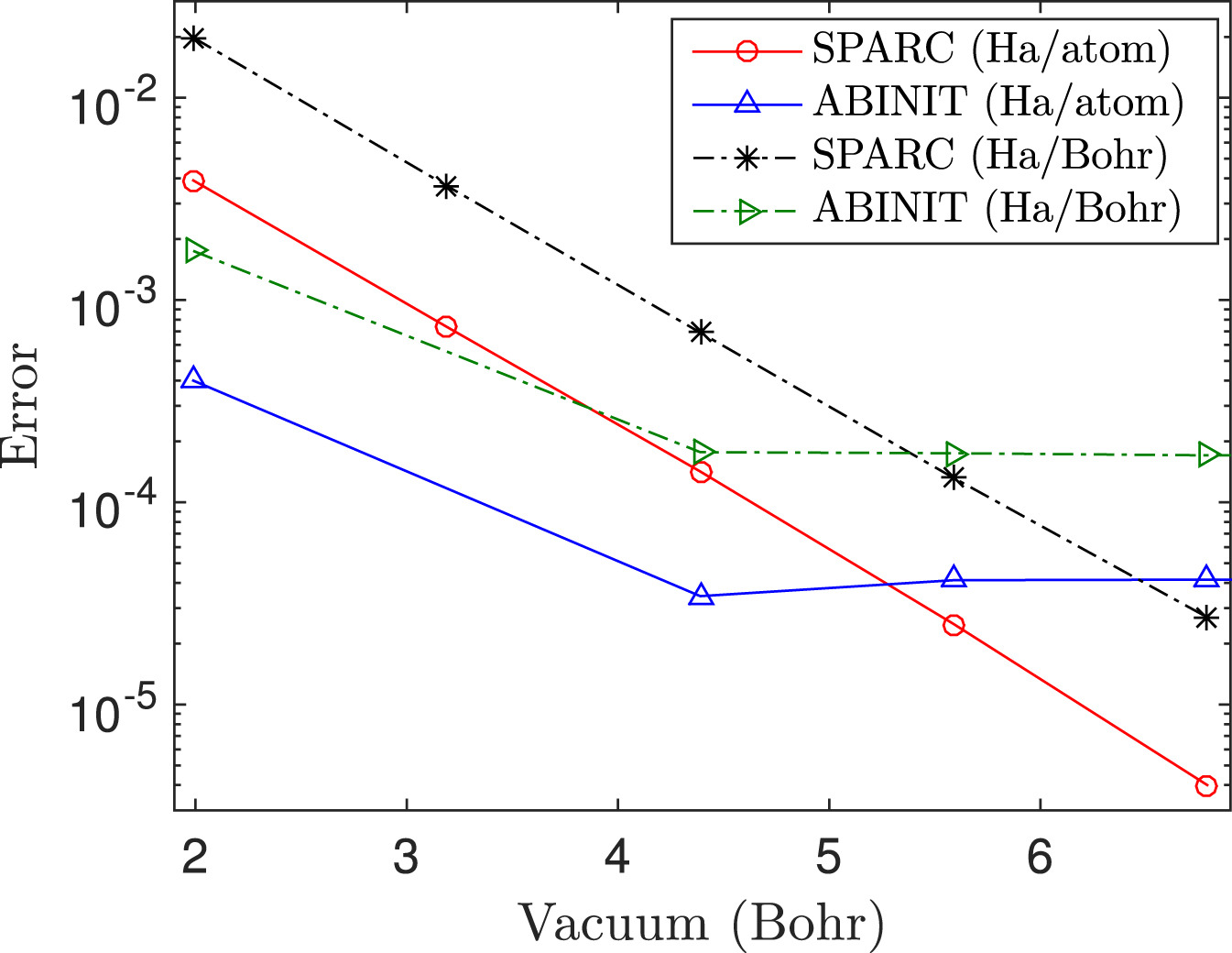
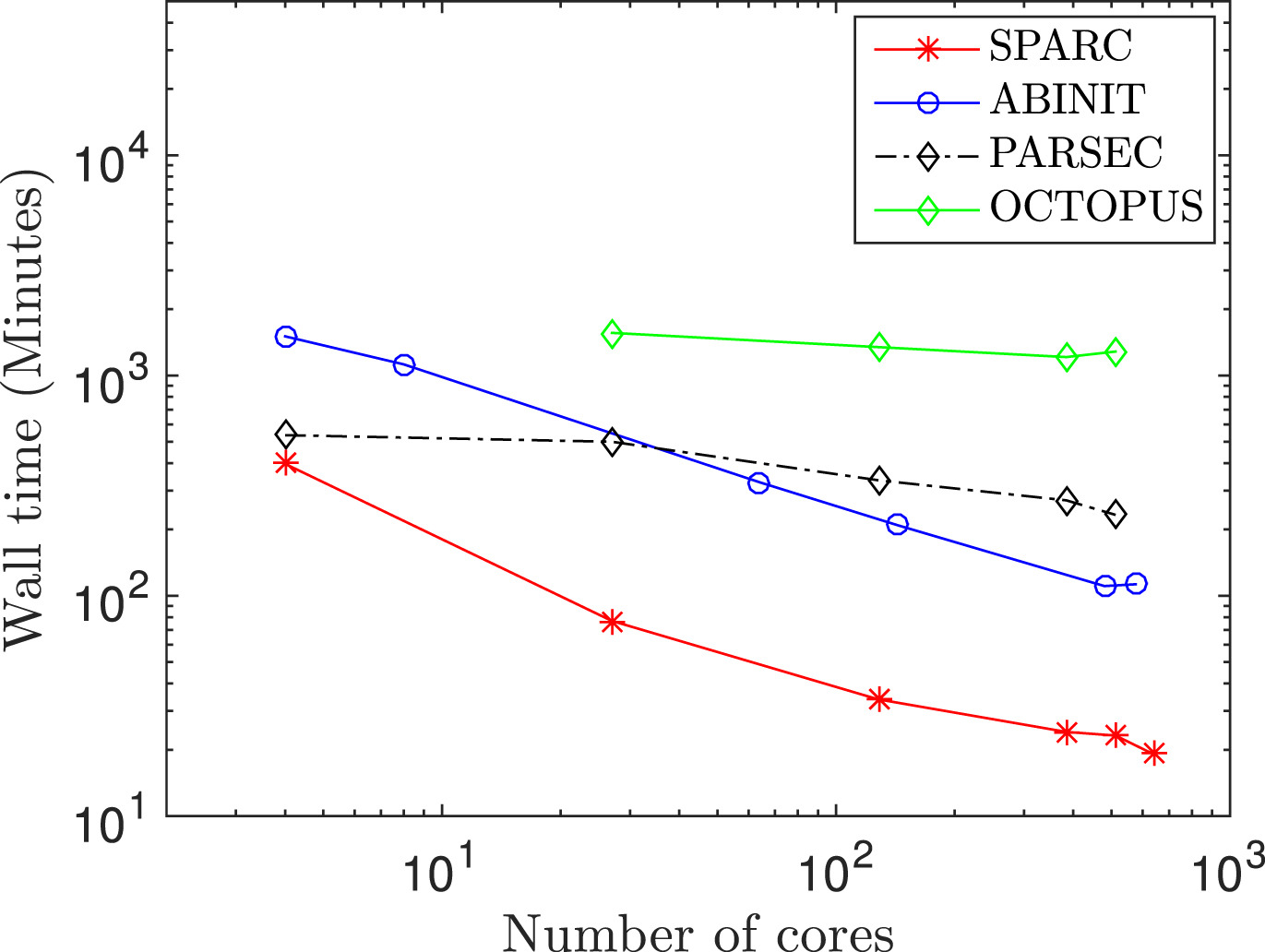

Traditional methods for DFT utilize the plane-wave basis. However, the non-locality of plane-waves makes them unsuitable for the development of approaches that scale linearly with respect to the number of atoms. Furthermore, developing parallel implementations that can efficiently utilize large-scale computer architectures is challenging. Finally, the need for periodic boundary conditions limits their effectiveness in the study of non-periodic and localized systems such as defects, as well as non-traditional symmetries like cyclic and helical. To overcome these limitations, we have developed a new real-space formulation and parallel implementation of DFT referred to as SPARC [SPARC1, SPARC2], which is highly competitive with plane-wave codes that have been developed by large teams of researchers over a couple of decades. SPARC has the potential to significantly impact a number of other fields including mechanics, materials science, physics, and chemistry, as evidenced by the fact that a large fraction of the world's computational resources are used for performing DFT calculations. Current efforts to extend the capabilities and performance of SPARC—the resulting framework is referred to as SPARC-X—with applications to catalysis, can be found here.